









单细胞测序技术是指在单细胞水平上,通过全基因组或转录组扩增,对核酸分子进行高通量测序的技术。该技术能够揭示单个细胞的基因结构和基因表达水平,反映细胞间的异质性,剖析单个细胞对生态系统或有机体的贡献[1,2]。1990 年,Iscove等[3]首次提出对单细胞进行转录组分析的构想,并用 PCR技术实现了对 cDNA 分子的指数级扩增。1992年,Telenius等[4]开发出寡核苷酸引物PCR(Degenerate oligonucleotide primed PCR,DOP-PCR)的方法,用简并寡核苷酸序列扩增基因组,为单细胞测序提供了思路。直到2001年,Dean等[5]首次使用随机六聚体引物和φ29 DNA聚合酶进行反应实现了DNA的滚环扩增,随后Raghunathan和Lasken等[6,7]于2005年发明了多重置换扩增技术(Multiple displacement amplification,MDA),实现了对单细胞全基因组的扩增与测序。但此时的单细胞扩增技术在覆盖度和扩增偏好性方面有明显的局限性,随后一些研究者致力于克服这些问题。例如,Stepanauskas等[8]于2017年使用φ29 DNA聚合酶的热稳定突变体提高了单细胞基因组测序的覆盖度;哈佛大学谢晓亮团队[9]于2012年发明了多次退火环状循环扩增技术(Multiple annealing and looping based amplification cycles,MALBAC),通过拟线性的扩增过程降低了指数扩增的序列偏好性。发展至今,单细胞测序技术在神经生物学、微生物学、胚胎发育、器官发生和免疫学研究中取得了广泛应用,临床上也已用于辅助生殖和肿瘤的诊断与治疗[10]。Nature Methods于2011年将单细胞研究方法列入最值得关注的技术领域,又于2013 年将相关应用列为年度最重要的方法学进展;近日Nature再次将单细胞测序技术评为2020年度最受期待的技术之一。
相比扩增子测序和宏基因组测序,单细胞扩增和测序技术有其独特的不可替代的优点。扩增子测序是指对微生物的特定基因进行测序[11],传统针对16S rDNA、18S rDNA或ITS(内转录间隔区)基因进行的扩增子测序虽然可以满足检测微生物群落多样性的需求,但这种方法很难准确鉴定到属以下的分类等级,也无法深入探究物种的功能信息[12]。宏基因组测序又称环境基因组测序或群落基因组测序,直接对样本中所有微生物的全基因组进行测序,可以同时对物种和功能基因做出鉴定,也有助于发掘潜在代谢途径。然而该方法容易忽视某些稀有种[13],且测序结果的组装也始终是一大难题[14,15]。如果说宏基因组数据集是捕获整个群落信息的一张巨网[16],那么单细胞测序方法则是分离目标基因组的“手术刀”和深入探究目标群落的“放大镜”,能不断细化、深化我们对微生物群落的认识[17]。
本综述重点介绍了在微生物生态学领域使用过或具有应用前景的单细胞分离技术和单细胞基因组扩增技术,并详细论述了单细胞测序技术在获得单细胞基因组、剖析种群异质性、探究种间关系、联合系统发育和功能信息等方面的应用,最后在此基础上对其在微生物生态学领域的发展做出了一些展望。
1 单细胞分离与全基因组扩增
通常单细胞测序的完整流程包括样品保存与制备、单细胞分离、细胞裂解、全基因组扩增(WGA)与产物的系统发育筛选、文库制备与测序、测序结果的质量控制[18],其中单细胞分离与全基因组扩增是最为关键的步骤,诸多技术已被开发并应用于微生物的相关领域,而如何根据科学问题对分离和扩增的方法进行选择或改进对科学研究具有重大意义。
1.1 细胞分离技术
细胞分离是单细胞测序的第一步,其准确性将直接影响后续的测序和分析。提高通量、减少样品与试剂的消耗、提高细胞分离捕获的灵敏度和精确性一直是研究者的目标。常用的细胞分离技术包括有限稀释法(Limited dilution)、显微操作法(Micromanipulation)、激光捕获显微分离技术(Laser capture microdissection)、拉曼镊子(Raman tweezers)、涡旋与相分隔(Vortex and phase-separation)、荧光激活细胞分选技术(Fluorescence-activated cell sorting,FACS)和微流控技术(Microfluidics)[19,20,21](表1)。其中微流控技术因其较低的成本、较高的通量和理想的分离效果在近10年发展迅速,成为细胞分离技术的主流方向[22]。
表1 常见单细胞分离技术一览
| 细胞分离技术 | 技术简介 | 优点 | 局限性 | 应用案例 |
|---|---|---|---|---|
| 有限稀释法 | 培养液稀释至约每0.1mL含有1个菌或细胞,根据泊松分布计算 | 简单廉价 | 准确性差,不具有针对性 | 对HIV-1病毒的全长序列进行测序[23] |
| 显微操作法 | 机械显微操作,用毛细管从各类样品中捕获单个细胞 | 对细胞进行可视性评估 | 通量极低,易对细胞造成机械损伤 | 分离并检测食物中的致病菌[24] |
| 激光捕获显微分离技术 | 将细胞进行固定和显色处理后直接分离 | 显色标记与分离结合,可分离复杂基质中的单细胞 | 通量低,易引入杂质或导致遗传信息丢失 | 研究动植物宿主-微生物的相互作用,鉴定未培养细菌,对单核原核细胞进行分析[25] |
| 拉曼镊子 | 通过拉曼显微镜区分生化特性不同的细胞,再用激光将其捕获 | 不需标记处理 | 只能分离生化特性显著不同的细胞 | 结合拉曼光镊和单细胞芯片分离癌细胞、红细胞、淋巴细胞和大肠杆菌[26] |
| 涡旋与相分隔 | 通过高速涡旋形成油包水体系 | 通量高,效率高,成本低 | 难以保证过程的精确性 | 用于探究硫酸盐还原功能类群[20] |
| 荧光细胞分选技术 | 基于所需细胞属性,结合多参数对特定细胞进行检测分离,同时评估生理学和分类学特性 | 灵敏度高,自动化程度高,可依据大小、粒度、荧光等属性分选 | 通量中等,成本高 | 分离丝状真菌并进行分类[27] |
| 微流控技术 | 通过微流控芯片原件等设备包裹和分离单细胞样品 | 通量高,精确稳定 | 需要特定仪器装备,开发周期较长 | 单细胞全基因组测序及抗性基因分析[21] |
1.2 单细胞基因扩增技术
图1
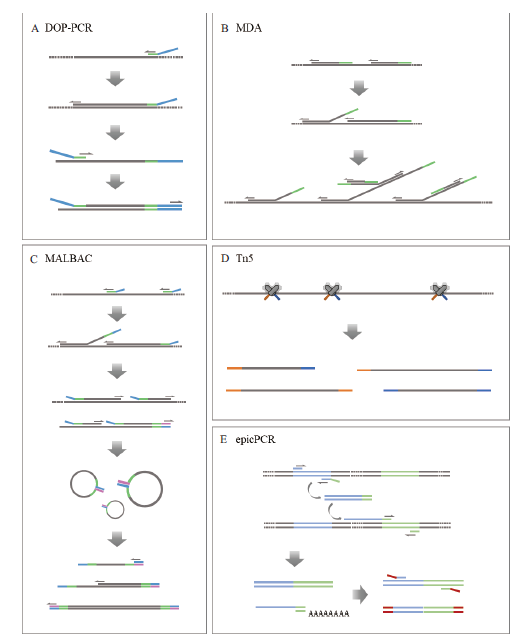
图1
常见单细胞基因组扩增技术原理示意图
A:简并寡核苷酸引物PCR技术(DOP-PCR)。随机引物的3' 端含6bp的随机序列,可以随机和基因组DNA结合,实现对全基因组的扩增;B:多重置换扩增技术(MDA)。随机六聚体引物与模板DNA结合,并在φ29 DNA聚合酶的作用下延伸;随后引物与延伸链结合,以多分支结构的形式延伸扩增;C:多次退火环状循环扩增技术(MALBAC)。首先引物与模板DNA结合,在具有置换活性的DNA聚合酶作用下延伸产生半扩增产物;随后随机引物与半扩增产物结合并延伸形成完整产物;最后对尾部互补成环的完整产物进行扩增;D:Tn5转座酶技术。Tn5转座酶随机将样品DNA片段化,并在小片段DNA两端加上特定的接头,便于后续的扩增和测序;E:细胞内融合基因技术(epicPCR)。两段目标基因被封装在同一微球中,在3条特殊引物的作用下产生融合片段;随后通过巢式PCR消除半扩增产物的影响,特异性扩增融合片段,并缩短其长度供二代测序
多重置换扩增技术(图1-B)是目前环境微生物领域最为成熟也是应用最广的单细胞基因组扩增技术。由于φ29 DNA聚合酶具有3'-5'核酸外切酶活性和校正活性,因此与DOP-PCR相比,多重置换扩增技术(Multiple displacement amplification,MDA)具有更高的序列覆盖度和保真度[30],但也存在外源DNA污染、序列覆盖不均、嵌合体干扰、序列组装与分析困难等不足[31]。由于MDA的偏差在一定程度上是随机的,所以通常可以通过多个数据集的混合拼接来减小这种偏差同时提高组装的完整性[32]。研究表明,2-5个单细胞扩增基因组数据集混合组装得到的基因组完整性中位数>97%,高于单个单细胞组装完整性的中位数(30%-90%)[33]。随后Povilaitis等[34]基于多重置换扩增的原理改进得到全基因组扩增技术WGA-X,使用耐热的突变体φ29 DNA聚合酶将延伸温度从30℃提高到45℃,大大提高了从CG含量高的单个环境细胞或病毒体回收基因组的能力。
多次退火环状循环扩增技术(Multiple annealing and looping-based amplification cycles,MALBAC)(图1-C)通过准线性预放大来减少与非线性放大相关的偏差。该技术利用特殊引物,使得扩增子的结尾互补而成环,从而很大程度上防止了DNA的指数性扩增[9]。MALBAC所使用的引物3'端是8个随机的核苷酸序列,5'端是27个固定的核苷酸序列,最大的特点在于它是准线性扩增而非指数扩增,因此拷贝数变异(Copy number variation,CNV)检测的准确性高且单核苷酸变异(Single nucleotide variants,SNV)检测的假阴性率低;而且,MALBAC的偏差具有可重复性,因此可进行降噪和归一化处理。然而,由于该技术使用的DNA聚合酶保真度低于φ29 DNA聚合酶,SNV检测的假阳性率高于MDA。目前MALBAC主要用于医疗诊断[35],在微生物单细胞的准确组装方面相比MDA优势不明显,在微生物生态学领域的应用前景不及MDA[36]。
Tn5转座酶(图1-D)最初用于二代测序的文库构建,将DNA片段化、末端修复、接头连接等简化为一步,大大简化建库步骤的同时为单细胞测序提供了有力工具。基于Tn5转座酶,谢晓亮团队[37]于2017年提出了改良的单细胞全基因组扩增方法(Linear amplification via transposon insertion,LIANTI),用Tn5转座子结合T7启动子形成的转座复合体随机插入单细胞基因组DNA,将基因组片段化并与T7启动子连接。随后T7启动子行使体外转录功能,用转录获得大量线性扩增的转录本,再经逆转录得到大量扩增产物,进行建库测序。该过程进行的是线性扩增,大大增强了扩增的稳定性,使LIANTI在遗传疾病的检测方面更加有效和精确。同年,Lan等[21]利用微流控技术将单细胞测序技术的通量提高到50 000个细胞/次,使得转座酶适用于环境基因组研究。
2 单细胞测序技术在微生态研究中的应用
目前,关于微生物生态的研究大多是群体水平,比如基于宏基因组学的研究能够获得群体的遗传潜力,尽管信息量很大,但无法探究个体的遗传潜力,细胞间的关联和异质性等问题也没有得到很好的解决[39]。单细胞测序与单细胞组学等新技术、新方法的出现为探索复杂环境中的单细胞生态学提供了崭新的机会[40]。单细胞基因组学的组装过程不依赖宏基因组数据的组装和分箱算法,因此能够较为准确地获得单个细胞内质粒、病毒等遗传信息[41],深入发掘不可培养的未知微生物的功能[42];该方法在个体细胞尺度上对样本进行研究[43],从而为探究种群异质性提供了有效手段。此外,单细胞基因组学也常用于研究基因之间的相互作用,破译微生物间或微生物与宿主的交流行为[44],揭示各种生态系统中微生物群落的结构和功能等[45]。
2.1 获得单细胞基因组
地球有丰富的微生物资源。根据比例法则结合生物多样性的对数正态模型进行的预测估计,全球微生物物种可能高达1012种[46]。其中大多数微生物是不可培养或还未被充分认识的,所以由于其未知性也被称作生态系统的暗物质[47]。无需培养的微生物分析鉴定方法(如宏基因组测序),自2004年出现以来一直广受欢迎[48]。然而,环境中微生物通常由个别丰度高的物种和大量丰度低的物种组成[49],由于采样方法、测序深度等限制,大多数研究都是针对丰度较高的种群开展的。Schlos等[50]对近11.3万个细菌和古菌OTU进行普查后发现环境中存在大量的稀有物种,且基于传统方法得到的信息非常有限。单细胞测序技术为获取稀有物种的基因组、发现新的代谢通路提供了支持[32]。
单细胞测序技术对细菌和古菌的暗物质基因组百科(GEBA-MDM)项目有很大贡献,推动了微生物的功能预测和系统发育鉴定[51],极大拓展了系统发育树上的微生物多样性[52]。2017年,Yu 等[53]基于微流控技术开发出一种具有单细胞分辨率的微型宏基因组方法,并用它分析了黄石国家公园的两个温泉样本,从中获得了29个新的基因组草图。Sieracki等[54]用单细胞基因组学方法研究了一系列海洋样品中小型原生生物的多样性,揭示了生态系统中新的相互作用和代谢途径。Ahrendt等[55]使用单细胞基因组学方法对8种不可培养真菌的基因组进行了测序,结果表明,通过结合多个扩增基因组,可以获得90%以上的单细胞基因组。
2.2 剖析种群异质性
Kashtan等[60]利用从1 000个原氯球菌细胞产生的大型单细胞扩增基因组文库来确定同一物种的不同生态型在整个季节变化中的基因组变异。测序结果显示,该种群由数百个具有不同“基因组骨架”(Genomic backbones)的亚种群组成,每个骨架包括一组不同的关键等位基因和一些特有的可变基因。Yoon等[61]对3个海洋原生生物(皮胆虫)进行单细胞鸟枪法测序,发现这些细胞代表了3种不同的微生态系,也为红藻亚界存在异养门提供了证据。Engel等[62]成功地应用单细胞基因组学评估了蜜蜂肠道微生物群中两个共生菌在物种水平上的异质性,揭示了菌株和生态位在代谢方面的特异性。2018年,Jochum等[63]对来自奥胡斯湾沉积物中的7个单细胞基因组进行了测序和分析,以了解它们在芳香化合物降解和能量代谢方面的潜力。研究证实了该种群具有代谢多样性,反映出微生物应对不同的能量条件和硫酸盐限制的生存策略。微生物所表现的这种种群异质性是一种适应性特征,可以提高微生物对多变或非均质环境条件的适应能力[64]。
2.3 探究种间关系
对微生物而言,每个细胞都是一个完整的遗传个体,包含了自身染色体、质粒、细胞器、病毒和其他内共生体的全部遗传信息。单细胞基因组学可以无差别地获得细胞内包含的所有遗传物质,从而在染色体DNA和其他遗传信息之间建立联系[65]。近五年的研究表明噬菌体与30多种细菌或古菌有关[66];还发现了一些能感染真核细胞的巨病毒[67]。2011年,Yoon等[61]在从海洋中分离出的单细胞中检测到了内生菌和病毒的遗传信息,揭示了单个细胞间的相互作用;于2014年,Roux等[68]结合单细胞测序和宏基因组测序取得了127个不可培养细菌的单细胞扩增基因组发现,SUP05病毒会使其宿主的能量代谢重构;2019年,Martinez-Hernandez等[69]发现远洋杆菌是海洋中数量极其丰富但未经培养的37-F6病毒的宿主;Labonte等[70]使用单细胞基因组学研究了扩散流、低温热液喷口中噬菌体与宿主的相互作用,结果表明,噬菌体感染后发生溶原化但不裂解宿主,与宿主共进化且增强了宿主对有毒金属和抗生素的抗性。
除此之外,单细胞测序也可用于探究细胞间的相互作用,发现微生物间的共生体,例如Nanoarchaeota和Ignicoccus的共生关系[71]、Actinomyces odontolyticus 和Candidatus Saccharibacteria 的寄生关系等[72]。低温透射电镜显示,酸性矿山废水中古菌之间常常存在物理性的胞间连接,例如细胞质桥、菌毛等[73]。如果胞间的相互作用足够强,不会在细胞分离过程中被破坏,单细胞测序技术则可将两个或多个细胞视作一个整体进行测序。Munson-McGee等[41]结合单细胞基因组测序和宏基因组测序发现,高温酸性温泉中专性共生纳米古菌与宿主的物理关联。2019年,Nakayama等[74]为了深入了解海洋蓝藻共生,对宿主远洋鞭毛藻进行单细胞测序并分析其中的蓝藻基因组。系统发育分析显示,样本中蓝藻属于新的分支,它与宿主鞭毛藻严格共存且经历了独立的进化,这种密切的共生关系导致它无法被传统宏基因组学检测到。因此,单细胞测序对发掘物种的多样性、生活方式、代谢途径和进化过程具有重大意义。
随着细胞内融合技术的发展成熟,2019年,Florenza等[38]建议使用epicPCR探究环境中真核生物和原核生物的相互作用关系,如固氮菌和浮游生物之间的互利共生[75],藻细胞周围的微型“生物圈”中有以碳水化合物为食的异养细菌[76],还有病原菌感染细胞并在宿主细胞中增殖[77]。该方法将单个真核细胞和伴生的原核细胞形成的单个细胞簇分散到液滴中,既实现了单个真核细胞的分隔,又保持了真核生物和相关原核生物之间紧密的联系。该方法也有一定局限性:只能揭示物理上紧密结合的相互作用,即在封装时紧密相连的相互作用,包括共生、捕食、寄生等,但无法区分相互作用关系发生在真核细胞的内部还是外部,也不能探究相互作用的分子机制。
2.4 联合系统发育和功能信息
2016年,Spencer等[20]首先开发了epicPCR技术,并将其用于硫酸盐还原细菌的研究,拓展了硫酸盐还原菌(Sulfate-reducing bacteria,SRB)的系统发育多样性。2019年,Qin等[78]用该技术对青藏高原盐湖沉积物中硫酸盐还原原核生物(Sulfate-reducing prokaryotes,SRPs)的系统发育进行了鉴定,研究表明西藏盐湖中有多种新的特有的SRP。随后,研究者将epicPCR技术用于抗性基因及其宿主的研究。由于抗性基因相对丰度较低且易在宿主间转移,传统的研究方法存在很大局限性,epicPCR技术的应用大大提高了抗性基因风险评估的精确度水平[79]。
2017年,Lan等[21]首先提出了结合微流控技术、片段化和序列标签的单细胞基因组测序Sic-seq,并使用这种方法来分析了环境样本中微生物耐药基因、毒力因子和噬菌体序列的分布。2020年,Chijiiwa等[80]发现借助单细胞基因组测序技术可以在没有参考基因组的情况下在物种水平上识别代谢应答者,为区分鉴定微生物群落中具有特定功能的未培养微生物提供了方法。Doud等[81]通过识别和捕获基于其原位功能或特征的单个微生物,扩充了功能驱动的单细胞基因组学。该方法先用荧光标记微生物含有的纤维素颗粒,然后通过荧光激活细胞分选方法分离单细胞进行宏基因组鸟枪法测序,结合16S rRNA基因扩增子测序进行系统发育定位,并对纤维素酶进行基因鉴定、表达检测和活性测试。这种功能驱动的单细胞方法可以将微生物分类直接与原位功能联系起来,具有广泛的应用前景和重大的实际意义。
3 总结与展望
2018年12月,英国皇家学会举办了一场以“单细胞生态学”为主题的跨学科会议,使用物理和分子领域相关的最新方法,在单细胞尺度研究生物现象,揭示同一物种的个体(或个体群)与其他个体、环境,以及不同种个体的相互作用[82]。操纵细胞的物理学家、研究微生物群落性质的微生物学家和开发新的单细胞方法的基因组学家齐聚一堂,产生了诸多新的见解与灵感。现在正是单细胞测序技术的飞速发展期,它的产生与完善推动了多个学科的进展,在各领域的广泛应用又促进了技术的成熟。目前已有一些较成熟的单细胞测序平台和商品化试剂盒上市,如10×genomic公司的10×Chromium Single Cell Gene Expression Solution 和Chromium Single Cell DNA Reagent Kits,BD公司的BD RhapsodyTM Single -Cell Analysis System,Wafergen公司的ICELL8 Single-Cell System,Bio-Rad公司的SureCell ATAC-Seq Library Preparation Kit和与Illumina合作开发的llumina® Bio-Rad® Single-Cell Sequencing Solution等。但由于单细胞测序的成本依然较高而能够同时检测的细胞数量也较为有限,其主要的应用热点依然以医学领域为主,环境微生物和微生物生态的应用才刚刚起步。而根据微生物生态研究的特点,领域内也有专家对单细胞测序的仪器使用方法进行了一系列优化,如结合单细胞测序技术将复杂群落分割成多个微型亚群再进行宏基因组测序的微型宏基因组(Mini-metagenome)技术[83],该技术降低了样品复杂性,同时具有传统宏基因组测序所不具备的单细胞分辨率,与单细胞技术相比又提高了测序通量,解决了嵌合体问题,非常适用于环境样本。单细胞测序技术有望成为微生物生态研究强大有力的主流技术力量,对于深入研究不可培养的微生物、加深对微生物生命之树的探索具有重大意义。
参考文献
单细胞测序技术及应用进展
[J].
Single cell sequencing technology and its applications progress
[J].
Single-cell genome sequencing:current state of the science
[J].
Haematopoiesis. Searching for stem cells
[J].
Degenerate oligonucleotide-primed PCR:General amplification of target DNA by a single degenerate primer
[J].
Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification
[J].
Genomic DNA amplification from a single bacterium
[J].
Single-cell genomic sequencing using Multiple Displacement Amplification.
[J].
Improved genome recovery and integrated cell-size analyses of individual uncultured microbial cells and viral particles
[J].
Genome-wide detection of single-nucleotide and copy-number variations of a single human cell
[J].
单细胞测序相关技术及其在生物医学研究中的应用
[J].
Development of single-cell sequencing and its biomedical applications
[J].
Next-generation sequencing of 16S ribosomal RNA gene amplicons
[J].
Analysis of the microbiome:Advantages of whole genome shotgun versus 16S amplicon sequencing
[J].
Sequencing depth and coverage:key considerations in genomic analyses
[J].
A user’s guide to quantitative and comparative analysis of metagenomic datasets
[J].
环境微生物宏基因组学研究中的生物信息学方法
[J].
Bioinformatics tools and applications in the study of environmental microbial metagenomics
[J].
Metagenomics-a guide from sampling to data analysis
[J].
DOI:10.1186/2042-5783-2-3
URL
PMID:22587947
[本文引用: 1]

Metagenomics applies a suite of genomic technologies and bioinformatics tools to directly access the genetic content of entire communities of organisms. The field of metagenomics has been responsible for substantial advances in microbial ecology, evolution, and diversity over the past 5 to 10 years, and many research laboratories are actively engaged in it now. With the growing numbers of activities also comes a plethora of methodological knowledge and expertise that should guide future developments in the field. This review summarizes the current opinions in metagenomics, and provides practical guidance and advice on sample processing, sequencing technology, assembly, binning, annotation, experimental design, statistical analysis, data storage, and data sharing. As more metagenomic datasets are generated, the availability of standardized procedures and shared data storage and analysis becomes increasingly important to ensure that output of individual projects can be assessed and compared.
Single cell genomics:an individual look at microbes
[J].
Sequencing of genomes from environmental single cells
[J].
DOI:10.1007/978-1-4939-7514-3_8
URL
PMID:29224071
[本文引用: 1]
Sequencing of single bacterial and archaeal cells is an important methodology that provides access to the genetic makeup of uncultivated microorganisms. We here describe the high-throughput fluorescence-activated cell sorting-based isolation of single cells from the environment, their lysis and strand displacement-mediated whole genome amplification. We further outline 16S rRNA gene sequence-based screening of single-cell amplification products, their preparation for Illumina sequencing libraries, and finally propose computational methods for read and contig level quality control of the resulting sequence data.
Single-cell analysis and isolation for microbiology and biotechnology:methods and applications
[J].
DOI:10.1007/s00253-010-2524-4
URL
PMID:20309540
[本文引用: 1]
Various single-cell isolation techniques, including dilution, micromanipulation, flow cytometry, microfluidics, and compartmentalization, have been developed. These techniques can be used to cultivate previously uncultured microbes, to assess and monitor cell physiology and function, and to screen for novel microbiological products. Various other techniques, such as viable staining, in situ hybridization, and those using autofluorescence proteins, are frequently combined with these single-cell isolation techniques depending on the purpose of the study. In this review article, we summarize currently available single-cell isolation techniques and their applications, when used in combination with other techniques, in microbiological and biotechnological studies.
Massively parallel sequencing of single cells by epicPCR links functional genes with phylogenetic markers
[J].
DOI:10.1038/ismej.2015.124
URL
PMID:26394010
[本文引用: 5]
Many microbial communities are characterized by high genetic diversity. 16S ribosomal RNA sequencing can determine community members, and metagenomics can determine the functional diversity, but resolving the functional role of individual cells in high throughput remains an unsolved challenge. Here, we describe epicPCR (Emulsion, Paired Isolation and Concatenation PCR), a new technique that links functional genes and phylogenetic markers in uncultured single cells, providing a throughput of hundreds of thousands of cells with costs comparable to one genomic library preparation. We demonstrate the utility of our technique in a natural environment by profiling a sulfate-reducing community in a freshwater lake, revealing both known sulfate reducers and discovering new putative sulfate reducers. Our method is adaptable to any conserved genetic trait and translates genetic associations from diverse microbial samples into a sequencing library that answers targeted ecological questions. Potential applications include identifying functional community members, tracing horizontal gene transfer networks and mapping ecological interactions between microbial cells.
Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding
[J].
DOI:10.1038/nbt.3880
URL
PMID:28553940
[本文引用: 4]
The application of single-cell genome sequencing to large cell populations has been hindered by technical challenges in isolating single cells during genome preparation. Here we present single-cell genomic sequencing (SiC-seq), which uses droplet microfluidics to isolate, fragment, and barcode the genomes of single cells, followed by Illumina sequencing of pooled DNA. We demonstrate ultra-high-throughput sequencing of >50,000 cells per run in a synthetic community of Gram-negative and Gram-positive bacteria and fungi. The sequenced genomes can be sorted in silico based on characteristic sequences. We use this approach to analyze the distributions of antibiotic-resistance genes, virulence factors, and phage sequences in microbial communities from an environmental sample. The ability to routinely sequence large populations of single cells will enable the de-convolution of genetic heterogeneity in diverse cell populations.
Recent advances in droplet-based microfluidic technologies for biochemistry and molecular biology
[J].
Single genome sequencing of near full-length HIV-1 RNA using a limiting dilution approach
[J].
Development of a micromanipulation method for single cell isolation of prokaryotes and its application in food safety
[J].
DOI:10.1371/journal.pone.0241865
URL
PMID:33152039
[本文引用: 1]
Research ethics has traditionally been guided by well-established documents such as the Belmont Report and the Declaration of Helsinki. At the same time, the introduction of Big Data methods, that is having a great impact in behavioral research, is raising complex ethical issues that make protection of research participants an increasingly difficult challenge. By conducting 39 semi-structured interviews with academic scholars in both Switzerland and United States, our research aims at exploring the code of ethics and research practices of academic scholars involved in Big Data studies in the fields of psychology and sociology to understand if the principles set by the Belmont Report are still considered relevant in Big Data research. Our study shows how scholars generally find traditional principles to be a suitable guide to perform ethical data research but, at the same time, they recognized and elaborated on the challenges embedded in their practical application. In addition, due to the growing introduction of new actors in scholarly research, such as data holders and owners, it was also questioned whether responsibility to protect research participants should fall solely on investigators. In order to appropriately address ethics issues in Big Data research projects, education in ethics, exchange and dialogue between research teams and scholars from different disciplines should be enhanced. In addition, models of consultancy and shared responsibility between investigators, data owners and review boards should be implemented in order to ensure better protection of research participants.
Laser microdissection:A promising tool for exploring microorganisms and their interactions with hosts
[J].
Nondestructive identification and accurate isolation of single cells through a chip with Raman optical tweezers
[J].
Flow cytometry and FACS applied to filamentous fungi
[J].
Single-cell whole-genome amplification and sequencing:Methodology and applications
[J].
DOI:10.1146/annurev-genom-090413-025352
URL
PMID:26077818
[本文引用: 1]
We present a survey of single-cell whole-genome amplification (WGA) methods, including degenerate oligonucleotide-primed polymerase chain reaction (DOP-PCR), multiple displacement amplification (MDA), and multiple annealing and looping-based amplification cycles (MALBAC). The key parameters to characterize the performance of these methods are defined, including genome coverage, uniformity, reproducibility, unmappable rates, chimera rates, allele dropout rates, false positive rates for calling single-nucleotide variations, and ability to call copy-number variations. Using these parameters, we compare five commercial WGA kits by performing deep sequencing of multiple single cells. We also discuss several major applications of single-cell genomics, including studies of whole-genome de novo mutation rates, the early evolution of cancer genomes, circulating tumor cells (CTCs), meiotic recombination of germ cells, preimplantation genetic diagnosis (PGD), and preimplantation genomic screening (PGS) for in vitro-fertilized embryos.
Improved DOP-PCR(iDOP-PCR):A robust and simple WGA method for efficient amplification of low copy number genomic DNA
[J].
Impact of whole genome amplification on analysis of copy number variants
[J].
DOI:10.1093/nar/gkn378
URL
PMID:18559357
[本文引用: 1]
Large-scale copy number variants (CNVs) have recently been recognized to play a role in human genome variation and disease. Approaches for analysis of CNVs in small samples such as microdissected tissues can be confounded by limited amounts of material. To facilitate analyses of such samples, whole genome amplification (WGA) techniques were developed. In this study, we explored the impact of Phi29 multiple-strand displacement amplification on detection of CNVs using oligonucleotide arrays. We extracted DNA from fresh frozen lymph node samples and used this for amplification and analysis on the Affymetrix Mapping 500k SNP array platform. We demonstrated that the WGA procedure introduces hundreds of potentially confounding CNV artifacts that can obscure detection of bona fide variants. Our analysis indicates that many artifacts are reproducible, and may correlate with proximity to chromosome ends and GC content. Pair-wise comparison of amplified products considerably reduced the number of apparent artifacts and partially restored the ability to detect real CNVs. Our results suggest WGA material may be appropriate for copy number analysis when amplified samples are compared to similarly amplified samples and that only the CNVs with the greatest significance values detected by such comparisons are likely to be representative of the unamplified samples.
Single-cell metagenomics:challenges and applications
[J].
The trajectory of microbial single-cell sequencing
[J].
Reconstructing each cell’s genome within complex microbial communities-dream or reality?
[J].
DOI:10.3389/fmicb.2014.00771
URL
PMID:25620966
[本文引用: 1]
As the vast majority of microorganisms have yet to be cultivated in a laboratory setting, access to their genetic makeup has largely been limited to cultivation-independent methods. These methods, namely metagenomics and more recently single-cell genomics, have become cornerstones for microbial ecology and environmental microbiology. One ultimate goal is the recovery of genome sequences from each cell within an environment to move toward a better understanding of community metabolic potential and to provide substrate for experimental work. As single-cell sequencing has the ability to decipher all sequence information contained in an individual cell, this method holds great promise in tackling such challenge. Methodological limitations and inherent biases however do exist, which will be discussed here based on environmental and benchmark data, to assess how far we are from reaching this goal.
In vitro evolution of phi29 DNA polymerase using isothermal compartmentalized self replication technique
[J].
Comparison of multiple displacement amplification(MDA)and multiple annealing and looping-based amplification cycles(MALBAC)in single-cell sequencing
[J].
A quantitative comparison of single-cell whole genome amplification methods
[J].
Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion(LIANTI)
[J].
Uncovering microbial inter-domain interactions in complex communities
[J].
Single-cell genomics and metagenomics for microbial diversity analysis
[M].
Nanoarchaeota, their Sulfolobales host, and Nanoarchaeota virus distribution across yellowstone national park hot springs
[J].
Function-driven single-cell genomics
[J].DOI:10.1111/1751-7915.12247 URL PMID:25627845 [本文引用: 1]
Defining cell types and states with single-cell genomics
[J].
Single-cell sequencing provides clues about the host interactions of segmented filamentous bacteria(SFB)
[J].
Genomic sequencing of uncultured microorganisms from single cells
[J].
DOI:10.1038/nrmicro2857
URL
PMID:22890147
[本文引用: 1]
Sequencing DNA from single cells has opened new windows onto the microbial world. It is becoming routine to sequence bacterial species directly from environmental samples or clinical specimens without the need to develop cultivation methods. Recent technical improvements often allow nearly complete genome assembly from these otherwise inaccessible species. New bioinformatics methods are also improving genome assembly from single cells. The use of single-cell sequencing in combination with metagenomic analysis is also emerging as a powerful new strategy to analyse bacterial communities. Here, the technical developments that have enabled single-cell sequencing, as well as some of the most exciting applications of this approach from the past few years, are reviewed.
Scaling laws predict global microbial diversity
[J].
DOI:10.1073/pnas.1521291113
URL
PMID:27140646
[本文引用: 1]
Scaling laws underpin unifying theories of biodiversity and are among the most predictively powerful relationships in biology. However, scaling laws developed for plants and animals often go untested or fail to hold for microorganisms. As a result, it is unclear whether scaling laws of biodiversity will span evolutionarily distant domains of life that encompass all modes of metabolism and scales of abundance. Using a global-scale compilation of approximately 35,000 sites and approximately 5.610(6) species, including the largest ever inventory of high-throughput molecular data and one of the largest compilations of plant and animal community data, we show similar rates of scaling in commonness and rarity across microorganisms and macroscopic plants and animals. We document a universal dominance scaling law that holds across 30 orders of magnitude, an unprecedented expanse that predicts the abundance of dominant ocean bacteria. In combining this scaling law with the lognormal model of biodiversity, we predict that Earth is home to upward of 1 trillion (10(12)) microbial species. Microbial biodiversity seems greater than ever anticipated yet predictable from the smallest to the largest microbiome.
Insights into the phylogeny and coding potential of microbial dark matter
[J].
Community structure and metabolism through reconstruction of microbial genomes from the environment
[J].
DOI:10.1038/nature02340
URL
PMID:14961025
[本文引用: 1]
Microbial communities are vital in the functioning of all ecosystems; however, most microorganisms are uncultivated, and their roles in natural systems are unclear. Here, using random shotgun sequencing of DNA from a natural acidophilic biofilm, we report reconstruction of near-complete genomes of Leptospirillum group II and Ferroplasma type II, and partial recovery of three other genomes. This was possible because the biofilm was dominated by a small number of species populations and the frequency of genomic rearrangements and gene insertions or deletions was relatively low. Because each sequence read came from a different individual, we could determine that single-nucleotide polymorphisms are the predominant form of heterogeneity at the strain level. The Leptospirillum group II genome had remarkably few nucleotide polymorphisms, despite the existence of low-abundance variants. The Ferroplasma type II genome seems to be a composite from three ancestral strains that have undergone homologous recombination to form a large population of mosaic genomes. Analysis of the gene complement for each organism revealed the pathways for carbon and nitrogen fixation and energy generation, and provided insights into survival strategies in an extreme environment.
Microbial diversity in the deep sea and the underexplored “rare biosphere”
[J].
DOI:10.1073/pnas.0605127103
URL
PMID:16880384
[本文引用: 1]
The evolution of marine microbes over billions of years predicts that the composition of microbial communities should be much greater than the published estimates of a few thousand distinct kinds of microbes per liter of seawater. By adopting a massively parallel tag sequencing strategy, we show that bacterial communities of deep water masses of the North Atlantic and diffuse flow hydrothermal vents are one to two orders of magnitude more complex than previously reported for any microbial environment. A relatively small number of different populations dominate all samples, but thousands of low-abundance populations account for most of the observed phylogenetic diversity. This
Status of the archaeal and bacterial census:an update
[J].
DOI:10.1128/mBio.00201-16
URL
PMID:27190214
[本文引用: 1]
UNLABELLED: A census is typically carried out for people across a range of geographical levels; however, microbial ecologists have implemented a molecular census of bacteria and archaea by sequencing their 16S rRNA genes. We assessed how well the census of full-length 16S rRNA gene sequences is proceeding in the context of recent advances in high-throughput sequencing technologies because full-length sequences are typically used as references for classification of the short sequences generated by newer technologies. Among the 1,411,234 and 53,546 full-length bacterial and archaeal sequences, 94.5% and 95.1% of the bacterial and archaeal sequences, respectively, belonged to operational taxonomic units (OTUs) that have been observed more than once. Although these metrics suggest that the census is approaching completion, 29.2% of the bacterial and 38.5% of the archaeal OTUs have been observed more than once. Thus, there is still considerable diversity to be explored. Unfortunately, the rate of new full-length sequences has been declining, and new sequences are primarily being deposited by a small number of studies. Furthermore, sequences from soil and aquatic environments, which are known to be rich in bacterial diversity, represent only 7.8 and 16.5% of the census, while sequences associated with host-associated environments represent 55.0% of the census. Continued use of traditional approaches and new technologies such as single-cell genomics and short-read assembly are likely to improve our ability to sample rare OTUs if it is possible to overcome this sampling bias. The success of ongoing efforts to use short-read sequencing to characterize archaeal and bacterial communities requires that researchers strive to expand the depth and breadth of this census. IMPORTANCE: The biodiversity contained within the bacterial and archaeal domains dwarfs that of the eukaryotes, and the services these organisms provide to the biosphere are critical. Surprisingly, we have done a relatively poor job of formally tracking the quality of the biodiversity as represented in full-length 16S rRNA genes. By understanding how this census is proceeding, it is possible to suggest the best allocation of resources for advancing the census. We found that the ongoing effort has done an excellent job of sampling the most abundant organisms but struggles to sample the rarer organisms. Through the use of new sequencing technologies, we should be able to obtain full-length sequences from these rare organisms. Furthermore, we suggest that by allocating more resources to sampling environments known to have the greatest biodiversity, we will be able to make significant advances in our characterization of archaeal and bacterial diversity.
A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea
[J].
DOI:10.1038/nature08656
URL
PMID:20033048
[本文引用: 1]
Sequencing of bacterial and archaeal genomes has revolutionized our understanding of the many roles played by microorganisms. There are now nearly 1,000 completed bacterial and archaeal genomes available, most of which were chosen for sequencing on the basis of their physiology. As a result, the perspective provided by the currently available genomes is limited by a highly biased phylogenetic distribution. To explore the value added by choosing microbial genomes for sequencing on the basis of their evolutionary relationships, we have sequenced and analysed the genomes of 56 culturable species of Bacteria and Archaea selected to maximize phylogenetic coverage. Analysis of these genomes demonstrated pronounced benefits (compared to an equivalent set of genomes randomly selected from the existing database) in diverse areas including the reconstruction of phylogenetic history, the discovery of new protein families and biological properties, and the prediction of functions for known genes from other organisms. Our results strongly support the need for systematic 'phylogenomic' efforts to compile a phylogeny-driven 'Genomic Encyclopedia of Bacteria and Archaea' in order to derive maximum knowledge from existing microbial genome data as well as from genome sequences to come.
Recent advances in genomic DNA sequencing of microbial species from single cells
[J].
DOI:10.1038/nrg3785
URL
PMID:25091868
[本文引用: 1]
The vast majority of microbial species remain uncultivated and, until recently, about half of all known bacterial phyla were identified only from their 16S ribosomal RNA gene sequence. With the advent of single-cell sequencing, genomes of uncultivated species are rapidly filling in unsequenced branches of the microbial phylogenetic tree. The wealth of new insights gained from these previously inaccessible groups is providing a deeper understanding of their basic biology, taxonomy and evolution, as well as their diverse roles in environmental ecosystems and human health.
Microfluidic-based mini-metagenomics enables discovery of novel microbial lineages from complex environmental samples
[J].
Single cell genomics yields a wide diversity of small planktonic protists across major ocean ecosystems
[J].
DOI:10.1038/s41598-019-42487-1
URL
PMID:30988337
[本文引用: 1]
Marine planktonic protists are critical components of ocean ecosystems and are highly diverse. Molecular sequencing methods are being used to describe this diversity and reveal new associations and metabolisms that are important to how these ecosystems function. We describe here the use of the single cell genomics approach to sample and interrogate the diversity of the smaller (pico- and nano-sized) protists from a range of oceanic samples. We created over 900 single amplified genomes (SAGs) from 8 Tara Ocean samples across the Indian Ocean and the Mediterranean Sea. We show that flow cytometric sorting of single cells effectively distinguishes plastidic and aplastidic cell types that agree with our understanding of protist phylogeny. Yields of genomic DNA with PCR-identifiable 18S rRNA gene sequence from single cells was low (15% of aplastidic cell sorts, and 7% of plastidic sorts) and tests with alternate primers and comparisons to metabarcoding did not reveal phylogenetic bias in the major protist groups. There was little evidence of significant bias against or in favor of any phylogenetic group expected or known to be present. The four open ocean stations in the Indian Ocean had similar communities, despite ranging from 14 degrees N to 20 degrees S latitude, and they differed from the Mediterranean station. Single cell genomics of protists suggests that the taxonomic diversity of the dominant taxa found in only several hundreds of microliters of surface seawater is similar to that found in molecular surveys where liters of sample are filtered.
Leveraging single-cell genomics to expand the fungal tree of life
[J].
基于16S rRNA和宏基因组高通量测序的微生物多样性研究
[D].
Microbial diversity research based on high-throughput sequencing data of 16S rRNA and metagenome
[D].
Bioinformatics challenges of new sequencing technology
[J].
DOI:10.1016/j.tig.2007.12.006
URL
PMID:18262676
[本文引用: 1]
New DNA sequencing technologies can sequence up to one billion bases in a single day at low cost, putting large-scale sequencing within the reach of many scientists. Many researchers are forging ahead with projects to sequence a range of species using the new technologies. However, these new technologies produce read lengths as short as 35-40 nucleotides, posing challenges for genome assembly and annotation. Here we review the challenges and describe some of the bioinformatics systems that are being proposed to solve them. We specifically address issues arising from using these technologies in assembly projects, both de novo and for resequencing purposes, as well as efforts to improve genome annotation in the fragmented assemblies produced by short read lengths.
基于三代测序技术的微生物组学研究进展
[J].
Analysis of prospective microbiology research using third-generation sequencing technology
[J].DOI:10.17520/biods.2018201 URL [本文引用: 1]
SMRT测序技术及其在微生物研究中的应用
[J].
SMRT Sequencing and Its application in microorganism studies
[J].
Single-cell genomics reveals hundreds of coexisting subpopulations in wild Prochlorococcus
[J].
DOI:10.1126/science.1248575
URL
PMID:24763590
[本文引用: 1]
Extensive genomic diversity within coexisting members of a microbial species has been revealed through selected cultured isolates and metagenomic assemblies. Yet, the cell-by-cell genomic composition of wild uncultured populations of co-occurring cells is largely unknown. In this work, we applied large-scale single-cell genomics to study populations of the globally abundant marine cyanobacterium Prochlorococcus. We show that they are composed of hundreds of subpopulations with distinct
Single-cell genomics reveals organismal interactions in uncultivated marine protists
[J].
DOI:10.1126/science.1203163
URL
PMID:21551060
[本文引用: 2]
Whole-genome shotgun sequence data from three individual cells isolated from seawater, followed by analysis of ribosomal DNA, indicated that the cells represented three divergent clades of picobiliphytes. In contrast with the recent description of this phylum, we found no evidence of plastid DNA nor of nuclear-encoded plastid-targeted proteins, which suggests that these picobiliphytes are heterotrophs. Genome data from one cell were dominated by sequences from a widespread single-stranded DNA virus. This virus was absent from the other two cells, both of which contained non-eukaryote DNA derived from marine Bacteroidetes and large DNA viruses. By using shotgun sequencing of uncultured marine picobiliphytes, we revealed the distinct interactions of individual cells.
Hidden diversity in honey bee gut symbionts detected by single-cell genomics
[J].
Single-cell genomics reveals a diverse metabolic potential of uncultivated Desulfatiglans-related deltaproteobacteria widely distributed in marine sediment
[J].
DOI:10.3389/fmicb.2018.02038
URL
PMID:30233524
[本文引用: 1]
Desulfatiglans-related organisms comprise one of the most abundant deltaproteobacterial lineages in marine sediments where they occur throughout the sediment column in a gradient of increasing sulfate and organic carbon limitation with depth. Characterized Desulfatiglans isolates are dissimilatory sulfate reducers able to grow by degrading aromatic hydrocarbons. The ecophysiology of environmental Desulfatiglans-populations is poorly understood, however, possibly utilization of aromatic compounds may explain their predominance in marine subsurface sediments. We sequenced and analyzed seven Desulfatiglans-related single-cell genomes (SAGs) from Aarhus Bay sediments to characterize their metabolic potential with regard to aromatic compound degradation and energy metabolism. The average genome assembly size was 1.3 Mbp and completeness estimates ranged between 20 and 50%. Five of the SAGs (group 1) originated from the sulfate-rich surface part of the sediment while two (group 2) originated from sulfate-depleted subsurface sediment. Based on 16S rRNA gene amplicon sequencing group 2 SAGs represent the more frequent types of Desulfatiglans-populations in Aarhus Bay sediments. Genes indicative of aromatic compound degradation could be identified in both groups, but the two groups were metabolically distinct with regard to energy conservation. Group 1 SAGs carry a full set of genes for dissimilatory sulfate reduction, whereas the group 2 SAGs lacked any genetic evidence for sulfate reduction. The latter may be due to incompleteness of the SAGs, but as alternative energy metabolisms group 2 SAGs carry the genetic potential for growth by acetogenesis and fermentation. Group 1 SAGs encoded reductive dehalogenase genes, allowing them to access organohalides and possibly conserve energy by their reduction. Both groups possess sulfatases unlike their cultured relatives allowing them to utilize sulfate esters as source of organic carbon and sulfate. In conclusion, the uncultivated marine Desulfatiglans populations are metabolically diverse, likely reflecting different strategies for coping with energy and sulfate limitation in the subsurface seabed.
Heterogeneity as an adaptive trait of microbial populations
[J].
Wiretapping into microbial interactions by single cell genomics
[J].DOI:10.3389/fmicb.2015.00258 URL PMID:25904902 [本文引用: 1]
Uncovering earth’s virome
[J].
DOI:10.1038/nature19094
URL
PMID:27533034
[本文引用: 1]
Viruses are the most abundant biological entities on Earth, but challenges in detecting, isolating, and classifying unknown viruses have prevented exhaustive surveys of the global virome. Here we analysed over 5 Tb of metagenomic sequence data from 3,042 geographically diverse samples to assess the global distribution, phylogenetic diversity, and host specificity of viruses. We discovered over 125,000 partial DNA viral genomes, including the largest phage yet identified, and increased the number of known viral genes by 16-fold. Half of the predicted partial viral genomes were clustered into genetically distinct groups, most of which included genes unrelated to those in known viruses. Using CRISPR spacers and transfer RNA matches to link viral groups to microbial host(s), we doubled the number of microbial phyla known to be infected by viruses, and identified viruses that can infect organisms from different phyla. Analysis of viral distribution across diverse ecosystems revealed strong habitat-type specificity for the vast majority of viruses, but also identified some cosmopolitan groups. Our results highlight an extensive global viral diversity and provide detailed insight into viral habitat distribution and host-virus interactions.
The rapidly expanding universe of giant viruses:Mimivirus, Pandoravirus, Pithovirus and Mollivirus
[J].
DOI:10.1093/femsre/fuv037
URL
PMID:26391910
[本文引用: 1]
More than a century ago, the term 'virus' was introduced to describe infectious agents that are invisible by light microscopy and capable of passing through sterilizing filters. In addition to their extremely small size, most viruses have minimal genomes and gene contents, and rely almost entirely on host cell-encoded functions to multiply. Unexpectedly, four different families of eukaryotic 'giant viruses' have been discovered over the past 10 years with genome sizes, gene contents and particle dimensions overlapping with that of cellular microbes. Their ongoing analyses are challenging accepted ideas about the diversity, evolution and origin of DNA viruses.
Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta- genomics
[J].
DOI:10.7554/eLife.03125
URL
PMID:25171894
[本文引用: 1]
Viruses modulate microbial communities and alter ecosystem functions. However, due to cultivation bottlenecks, specific virus-host interaction dynamics remain cryptic. In this study, we examined 127 single-cell amplified genomes (SAGs) from uncultivated SUP05 bacteria isolated from a model marine oxygen minimum zone (OMZ) to identify 69 viral contigs representing five new genera within dsDNA Caudovirales and ssDNA Microviridae. Infection frequencies suggest that approximately 1/3 of SUP05 bacteria is viral-infected, with higher infection frequency where oxygen-deficiency was most severe. Observed Microviridae clonality suggests recovery of bloom-terminating viruses, while systematic co-infection between dsDNA and ssDNA viruses posits previously unrecognized cooperation modes. Analyses of 186 microbial and viral metagenomes revealed that SUP05 viruses persisted for years, but remained endemic to the OMZ. Finally, identification of virus-encoded dissimilatory sulfite reductase suggests SUP05 viruses reprogram their host's energy metabolism. Together, these results demonstrate closely coupled SUP05 virus-host co-evolutionary dynamics with the potential to modulate biogeochemical cycling in climate-critical and expanding OMZs.
Single-cell genomics uncover Pelagibacter as the putative host of the extremely abundant uncultured 37-F6 viral population in the ocean
[J].
Single cell genomics-based analysis of gene content and expression of prophages in a diffuse-flow deep-sea hydrothermal system
[J].
A new phylum of Archaea represented by a nanosized hyperthermophilic symbiont
[J].
DOI:10.1038/417063a
URL
PMID:11986665
[本文引用: 1]
According to small subunit ribosomal RNA (ss rRNA) sequence comparisons all known Archaea belong to the phyla Crenarchaeota, Euryarchaeota, and--indicated only by environmental DNA sequences--to the 'Korarchaeota'. Here we report the cultivation of a new nanosized hyperthermophilic archaeon from a submarine hot vent. This archaeon cannot be attached to one of these groups and therefore must represent an unknown phylum which we name 'Nanoarchaeota' and species, which we name 'Nanoarchaeum equitans'. Cells of 'N. equitans' are spherical, and only about 400 nm in diameter. They grow attached to the surface of a specific archaeal host, a new member of the genus Ignicoccus. The distribution of the 'Nanoarchaeota' is so far unknown. Owing to their unusual ss rRNA sequence, members remained undetectable by commonly used ecological studies based on the polymerase chain reaction. 'N. equitans' harbours the smallest archaeal genome; it is only 0.5 megabases in size. This organism will provide insight into the evolution of thermophily, of tiny genomes and of interspecies communication.
Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle
[J].
Inter-species interconnections in acid mine drainage microbial communities
[J].
Single-cell genomics unveiled a cryptic cyanobacterial lineage with a worldwide distribution hidden by a dinoflagellate host
[J].
DOI:10.1073/pnas.1902538116
URL
PMID:31235587
[本文引用: 1]
Cyanobacteria are one of the most important contributors to oceanic primary production and survive in a wide range of marine habitats. Much effort has been made to understand their ecological features, diversity, and evolution, based mainly on data from free-living cyanobacterial species. In addition, symbiosis has emerged as an important lifestyle of oceanic microbes and increasing knowledge of cyanobacteria in symbiotic relationships with unicellular eukaryotes suggests their significance in understanding the global oceanic ecosystem. However, detailed characteristics of these cyanobacteria remain poorly described. To gain better insight into marine cyanobacteria in symbiosis, we sequenced the genome of cyanobacteria collected from a cell of a pelagic dinoflagellate that is known to host cyanobacterial symbionts within a specialized chamber. Phylogenetic analyses using the genome sequence revealed that the cyanobacterium represents an underdescribed lineage within an extensively studied, ecologically important group of marine cyanobacteria. Metagenomic analyses demonstrated that this cyanobacterial lineage is globally distributed and strictly coexists with its host dinoflagellates, suggesting that the intimate symbiotic association allowed the cyanobacteria to escape from previous metagenomic studies. Furthermore, a comparative analysis of the protein repertoire with related species indicated that the lineage has independently undergone reductive genome evolution to a similar extent as Prochlorococcus, which has the most reduced genomes among free-living cyanobacteria. Discovery of this cyanobacterial lineage, hidden by its symbiotic lifestyle, provides crucial insights into the diversity, ecology, and evolution of marine cyanobacteria and suggests the existence of other undiscovered cryptic cyanobacterial lineages.
Identification of associations between bacterioplankton and photosynthetic picoeukaryotes in coastal waters
[J].
Zooming in on the phycosphere:the ecological interface for phytoplankton-bacteria relationships
[J].
Intracellular survival and replication of Vibrio cholerae O139 in aquatic free-living amoebae
[J].
DOI:10.1111/j.1462-2920.2005.00771.x
URL
PMID:15946296
[本文引用: 1]
Vibrio cholerae is a highly infectious bacterium responsible for large outbreaks of cholera among humans at regular intervals. A seasonal distribution of epidemics is known but the role of naturally occurring habitats are virtually unknown. Plankton has been suggested to play a role, because bacteria can attach to such organisms forming a biofilm. Acanthamoebea castellanii is an environmental amoeba that has been shown to be able to ingest and promote growth of several bacteria of different origin. The aim of the present study was to determine whether or not an intra-amoebic behaviour of V. cholerae O139 exists. Interaction between these microorganisms in co-culture was studied by culturable counts, gentamicin assay, electron microscopy, and polymerase chain reaction. The interaction resulted in intra-amoebic growth and survival of V. cholerae in the cytoplasm of trophozoites as well as in the cysts of A. castellanii. These data show symbiosis between these microorganisms, a facultative intracellular behaviour of V. cholerae contradicting the generally held view, and a role of free-living amoebae as hosts for V. cholerae O139. Taken together, this opens new doors to study the ecology, immunity, epidemiology, and treatment of cholera.
Unraveling the diversity of sedimentary sulfate-reducing prokaryotes(SRP)across Tibetan saline lakes using epicPCR
[J].
Host range of antibiotic resistance genes in wastewater treatment plant influent and effluent
[J].DOI:10.1093/femsle/fnaa156 URL PMID:33068111 [本文引用: 1]
Single-cell genomics of uncultured bacteria reveals dietary fiber responders in the mouse gut microbiota
[J].
Function-driven single-cell genomics uncovers cellulose-degrading bacteria from the rare biosphere
[J].DOI:10.1038/s41396-019-0557-y URL PMID:31754206 [本文引用: 1]
Single cell ecology
[J].

{kind=link}
{kind=link}