







生物技术通报 ›› 2021, Vol. 37 ›› Issue (8): 131-140.doi: 10.13560/j.cnki.biotech.bull.1985.2021-0191
尚骁尧( ), 周玲芳, 尹芊芊, 晁跃辉()
), 周玲芳, 尹芊芊, 晁跃辉()
收稿日期:2021-02-18
出版日期:2021-08-26
发布日期:2021-09-10
作者简介:尚骁尧,女,硕士研究生,研究方向:草地植物生物技术;E-mail: 基金资助:
SHANG Xiao-yao(), ZHOU Ling-fang, YIN Qian-qian, CHAO Yue-hui()
Received:2021-02-18
Published:2021-08-26
Online:2021-09-10
摘要:
为了深入分析和探索豆科模式植物蒺藜苜蓿的mRNA完整结构,使用单分子长读数测序技术(single-molecule long-read sequencing technology,SMRT)对蒺藜苜蓿进行全长转录组测序及分析。共获得7 728 183个subread和509 014条全长非嵌合序列(full-length non-chimeric read,FLNC),通过比对分析发现,94.36%的序列与93.01%的序列分别与蒺藜苜蓿R108与A17参考基因组匹配。总计存在8 406种可变性剪接,其中主要的剪接方式为内含子保留(intron retention,RI)。共发现23 926个基因,其中12 049个基因存在295 545条转录本,在这些转录本中至少存在一个poly(A)位点。此外,共鉴定出3 223条转录因子,6 595条长非编码RNA(long non-coding RNA,lncRNA)和479条融合转录本。使用SMRT技术能够深入发掘蒺藜苜蓿转录数据,也为更好地利用蒺藜苜蓿基因组资源提供数据补充。
尚骁尧, 周玲芳, 尹芊芊, 晁跃辉. 蒺藜苜蓿(Medicago truncatula)全长转录组测序及分析[J]. 生物技术通报, 2021, 37(8): 131-140.
SHANG Xiao-yao, ZHOU Ling-fang, YIN Qian-qian, CHAO Yue-hui. Sequencing and Analysis of Full-length Transcriptome from Medicago truncatula[J]. Biotechnology Bulletin, 2021, 37(8): 131-140.
| 类别 Category | 数目 Amount | 最小长度 Min length/bp | 最大长度 Max length/bp | 平均长度 Mean length/bp | N50长度 N50 length/bp |
|---|---|---|---|---|---|
| Subread | 7 728 183 | 51 | 73 164 | 1 524 | 2 051 |
| CCS | 659 220 | 200 | 17 791 | 2 257 | 2 757 |
| FLNC | 509 014 | 200 | 17 128 | 2 047 | 2 440 |
| ICE consensus | 243 832 | 200 | 17 128 | 2 305 | 3 367 |
| Polished consensus | 243 676 | 166 | 17 128 | 2 321 | 3 380 |
| Corrected consensus | 236 243 | 260 | 17 954 | 2 425 | 3 496 |
表1 SMRT测序数据统计
Table 1 Statistics of sequencing data by SMRT
| 类别 Category | 数目 Amount | 最小长度 Min length/bp | 最大长度 Max length/bp | 平均长度 Mean length/bp | N50长度 N50 length/bp |
|---|---|---|---|---|---|
| Subread | 7 728 183 | 51 | 73 164 | 1 524 | 2 051 |
| CCS | 659 220 | 200 | 17 791 | 2 257 | 2 757 |
| FLNC | 509 014 | 200 | 17 128 | 2 047 | 2 440 |
| ICE consensus | 243 832 | 200 | 17 128 | 2 305 | 3 367 |
| Polished consensus | 243 676 | 166 | 17 128 | 2 321 | 3 380 |
| Corrected consensus | 236 243 | 260 | 17 954 | 2 425 | 3 496 |
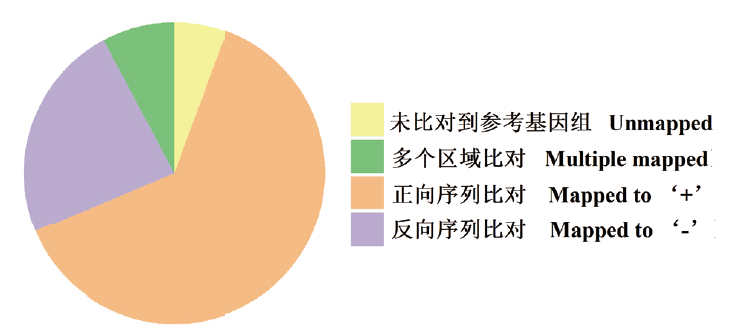
图1 校正后序列与参考基因组比对分析
Fig. 1 GMAP analysis of corrected reads to reference genome
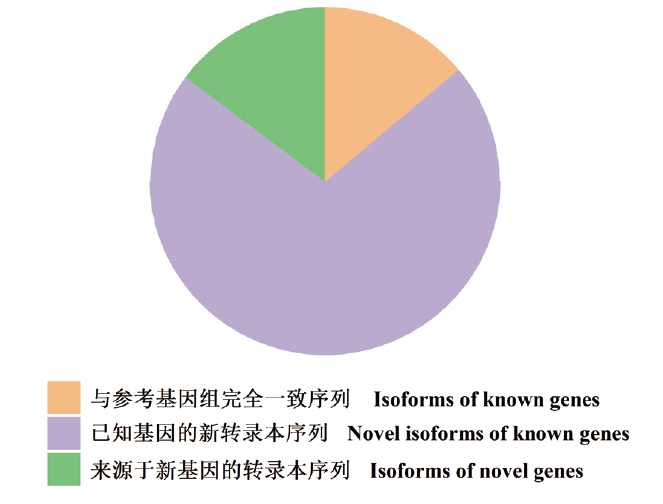
图2 RC61125序列的分类
Fig. 2 Classification of RC61125 sequence
| 数据库 Databases | 新基因注释数量 Number of functional annotated novel genes |
|---|---|
| NR | 5 183 |
| SwissProt | 2 370 |
| KEGG | 4 532 |
| KOG | 1 884 |
| GO | 1 717 |
| NT | 6 916 |
| PFAM | 1 717 |
| 总计 Total | 7 209 |
| 未注释 Unannotated ones | 227 |
表2 新基因功能注释
Table 2 Functional annotations of novel genes
| 数据库 Databases | 新基因注释数量 Number of functional annotated novel genes |
|---|---|
| NR | 5 183 |
| SwissProt | 2 370 |
| KEGG | 4 532 |
| KOG | 1 884 |
| GO | 1 717 |
| NT | 6 916 |
| PFAM | 1 717 |
| 总计 Total | 7 209 |
| 未注释 Unannotated ones | 227 |

图3 新基因Nr同源物种分布图
Fig. 3 Nr homologous species distribution diagram of novel genes
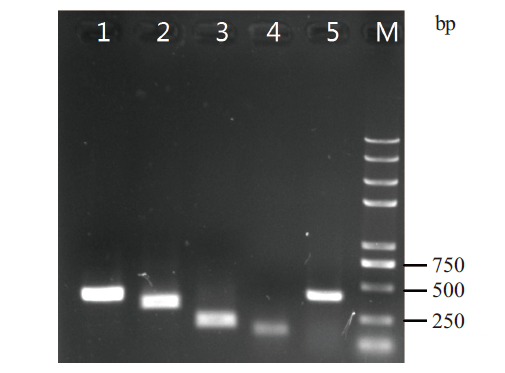
图4 新基因RT-PCR验证 1-5:随机挑选新基因;M:DNA marker
Fig. 4 RT-PCR validation of novel genes 1-5:Randomly selected novel genes. M:DNA marker
| 序列来源 Sequence origin | 编码区数量 Number of CDS | 编码区平均长度 Mean CDS length/bp | 完整编码区数量 Number of complete CDS | 3'非编码区数量 Number of 3'-UTR | 5'非编码区数量 Number of 5'-UTR |
|---|---|---|---|---|---|
| SMRT | 36 235 | 1 027.03 | 13 451 | 1 990 | 11 071 |
| R108 | 61 020 | 874.54 | 16 313 | 3 895 | 4 928 |
表3 编码区及非编码区鉴定
Table 3 CDS and UTR identification
| 序列来源 Sequence origin | 编码区数量 Number of CDS | 编码区平均长度 Mean CDS length/bp | 完整编码区数量 Number of complete CDS | 3'非编码区数量 Number of 3'-UTR | 5'非编码区数量 Number of 5'-UTR |
|---|---|---|---|---|---|
| SMRT | 36 235 | 1 027.03 | 13 451 | 1 990 | 11 071 |
| R108 | 61 020 | 874.54 | 16 313 | 3 895 | 4 928 |

图5 转录本外显子分布 Pac-Bio:SMRT测序数据;R108:蒺藜苜蓿R108参考基因组数据;A17:蒺藜苜蓿A17参考基因组数据
Fig.5 Distribution of exons in transcripts Pac-Bio:SMRT transcripts. R108:Transcripts from M. truncatula R108 reference genome.A17:Transcripts from M. truncatula A17 reference genome

图6 AS数量及类型 SE:外显子跳跃;MX:外显子互斥;A5:可变性5'剪切位点;A3:可变性3'剪切位点;RI:内含子保留;AF:第一外显子可变剪接;AL:最后外显子可变剪接
Fig.6 Numbers and types of AS events SE:Skipped exon. MX:mutually exclusive exon. A5:Alternative 5' splice site. A3:Alternative 3' splice site. RI:Retained intron. AF:First exon is alternative splice. AL:Last exon is alternative splice
| 转录本数目 Number of transcript | SMRT测序 SMRT sequence | 参考基因组 Reference genome |
|---|---|---|
| 1 | 11 730 | 52 332 |
| 2 | 4 713 | 2 308 |
| 3 | 2 660 | 617 |
| 4 | 1 614 | 246 |
| 5 | 965 | 96 |
| 6 | 618 | 58 |
| 7 | 415 | 21 |
| 8 | 334 | 10 |
| 9 | 216 | 8 |
| 10 | 147 | 5 |
| >10 | 514 | 5 |
表4 SMRT测序及参考基因组转录本数目
Table 4 Number of transcripts in SMRT sequence and reference genomes
| 转录本数目 Number of transcript | SMRT测序 SMRT sequence | 参考基因组 Reference genome |
|---|---|---|
| 1 | 11 730 | 52 332 |
| 2 | 4 713 | 2 308 |
| 3 | 2 660 | 617 |
| 4 | 1 614 | 246 |
| 5 | 965 | 96 |
| 6 | 618 | 58 |
| 7 | 415 | 21 |
| 8 | 334 | 10 |
| 9 | 216 | 8 |
| 10 | 147 | 5 |
| >10 | 514 | 5 |

图7 lncRNA鉴定 A:使用CPC、CNCI、PLEK及Pfam 4种方法预测lncRNA韦恩图;B:SMRT鉴定的:4种lncRNA类型比例
Fig.7 Identification of lncRNA A:Venn diagram of lncRNAs predicted by CPC,CNCI,PLEK and Pfam methods.B:Proportions of 4 types of lncRNA identified by SMRT
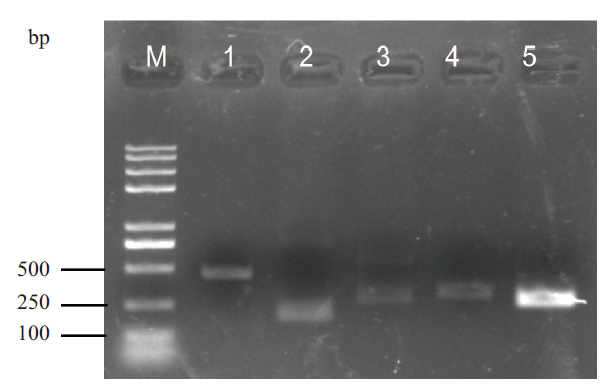
图8 融合转录本RT-PCR验证 1-5:随机挑选融合转录本;M:DNA marker
Fig. 8 RT-PCR validation of fusion transcripts 1-5:Randomly selected fusion transcripts. M:DNA marker

图9 poly(A)位点分析 A:基因poly(A)位点数目分布;B:poly(A)剪切位点附近核苷酸组成;C:poly(A)位点上游重复序列MEME分析;D:poly(A)位点下游游重复序列MEME分析
Fig. 9 Analysis of poly(A)sites A:Distribution of the number of poly(A)sites per gene. B:Nucleotide composition around poly(A)cleavage sites. C:MEME analysis of repeated sequence in the upstream of the poly(A)site. D:MEME analysis of repeated sequence in the downstream of the poly(A)site
| [1] |
Young ND, Debellé F, Oldroyd GE, et al. The Medicago genome provides insight into the evolution of rhizobial symbioses[J]. Nature, 2011, 480(7378):520-524.
doi: 10.1038/nature10625 URL |
| [2] |
Tang H, Krishnakumar V, Bidwell S, et al. An improved genome release(version Mt4. 0)for the model legume Medicago truncatula[J]. BMC Genomics, 2014, 15:312.
doi: 10.1186/1471-2164-15-312 URL |
| [3] |
Tadege M, Ratet P, Mysore KS. Insertional mutagenesis:a Swiss Army knife for functional genomics of Medicago truncatula[J]. Trends Plant Sci, 2005, 10(5):229-235.
doi: 10.1016/j.tplants.2005.03.009 URL |
| [4] |
Tadege M, Wen JQ, He J, et al. Large-scale insertional mutagenesis using the Tnt1 retrotransposon in the model legume Medicago truncatula[J]. Plant J, 2008, 54(2):335-347.
doi: 10.1111/j.1365-313X.2008.03418.x URL |
| [5] |
Donà M, Confalonieri M, Minio A, et al. RNA-Seq analysis discloses early senescence and nucleolar dysfunction triggered by Tdp1α depletion in Medicago truncatula[J]. J Exp Bot, 2013, 64(7):1941-1951.
doi: 10.1093/jxb/ert063 URL |
| [6] |
Cabeza RA, Liese R, Lingner A, et al. RNA-seq transcriptome profiling reveals that Medicago truncatula nodules acclimate N₂ fixation before emerging P deficiency reaches the nodules[J]. J Exp Bot, 2014, 65(20):6035-6048.
doi: 10.1093/jxb/eru341 URL |
| [7] |
Thatcher LF, Williams AH, Garg G, et al. Transcriptome analysis of the fungal pathogen Fusarium oxysporum f. sp. medicaginis during colonisation of resistant and susceptible Medicago truncatula hosts identifies differential pathogenicity profiles and novel candidate effectors[J]. BMC Genomics, 2016, 17(1):860.
pmid: 27809762 |
| [8] |
Reddy ASN, Marquez Y, Kalyna M, et al. Complexity of the alternative splicing landscape in plants[J]. The Plant cell, 2013, 25(10):3657-3683.
doi: 10.1105/tpc.113.117523 URL |
| [9] |
Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation:extent, regulation and function[J]. Nat Rev Genet, 2013, 14(7):496-506.
doi: 10.1038/nrg3482 pmid: 23774734 |
| [10] |
Zhu FY, Chen MX, Ye NH, et al. Proteogenomic analysis reveals alternative splicing and translation as part of the abscisic acid response in Arabidopsis seedlings[J]. Plant J, 2017, 91(3):518-533.
doi: 10.1111/tpj.13571 URL |
| [11] |
Zhang P, Deng H, Xiao FM, et al. Alterations of alternative splicing patterns of Ser/arg-rich(SR)genes in response to hormones and stresses treatments in different ecotypes of rice(Oryza sativa)[J]. J Integr Agric, 2013, 12(5):737-748.
doi: 10.1016/S2095-3119(13)60260-9 URL |
| [12] |
Rodet F, Lelong C, Dubos MP, et al. Alternative splicing of a single precursor mRNA generates two subtypes of Gonadotropin-Releasing Hormone receptor orthologues and their variants in the bivalve mollusc Crassostrea gigas[J]. Gene, 2008, 414(1/2):1-9.
doi: 10.1016/j.gene.2008.01.022 URL |
| [13] |
Palusa SG, Ali GS, Reddy AS. Alternative splicing of pre-mRNAs of Arabidopsis serine/arginine-rich proteins:regulation by hormones and stresses[J]. Plant J, 2007, 49(6):1091-1107.
doi: 10.1111/tpj.2007.49.issue-6 URL |
| [14] |
Dowhan DH, Hong EP, Auboeuf D, et al. Steroid hormone receptor coactivation and alternative RNA splicing by U2AF65-related proteins CAPERα and CAPERβ[J]. Mol Cell, 2005, 17(3):429-439.
doi: 10.1016/j.molcel.2004.12.025 URL |
| [15] |
Au KF, Sebastiano V, Afshar PT, et al. Characterization of the human ESC transcriptome by hybrid sequencing[J]. PNAS, 2013, 110(50):E4821-E4830.
doi: 10.1073/pnas.1320101110 URL |
| [16] |
Gordon SP, Tseng E, Salamov A, et al. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing[J]. PLoS One, 2015, 10(7):e0132628.
doi: 10.1371/journal.pone.0132628 URL |
| [17] |
Sharon D, Tilgner H, Grubert F, et al. A single-molecule long-read survey of the human transcriptome[J]. Nat Biotechnol, 2013, 31(11):1009-1014.
doi: 10.1038/nbt.2705 URL |
| [18] |
Dong L, Liu H, Zhang J, et al. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research[J]. BMC Genomics, 2015, 16:1039.
doi: 10.1186/s12864-015-2257-y URL |
| [19] |
Wang B, Tseng E, Regulski M, et al. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing[J]. Nat Commun, 2016, 7:11708.
doi: 10.1038/ncomms11708 pmid: 27339440 |
| [20] |
Chao YH, Yuan JB, Li SF, et al. Analysis of transcripts and splice isoforms in red clover(Trifolium pratense L.)by single-molecule long-read sequencing[J]. BMC Plant Biol, 2018, 18(1):300.
doi: 10.1186/s12870-018-1534-8 URL |
| [21] |
Abdel-Ghany SE, Hamilton M, Jacobi JL, et al. A survey of the Sorghum transcriptome using single-molecule long reads[J]. Nat Commun, 2016, 7:11706.
doi: 10.1038/ncomms11706 pmid: 27339290 |
| [22] |
Alamancos GP, Pagès A, Trincado JL, et al. Leveraging transcript quantification for fast computation of alternative splicing profiles[J]. RNA, 2015, 21(9):1521-1531.
doi: 10.1261/rna.051557.115 pmid: 26179515 |
| [23] |
Shimizu K, Adachi J, Muraoka Y. ANGLE:a sequencing errors resistant program for predicting protein coding regions in unfinished cDNA[J]. J Bioinform Comput Biol, 2006, 4(3):649-664.
doi: 10.1142/S0219720006002260 URL |
| [24] |
Kong L, Zhang Y, Ye ZQ, et al. CPC:assess the protein-coding potential of transcripts using sequence features and support vector machine[J]. Nucleic Acids Res, 2007, 35(web server issue):W345-W349.
doi: 10.1093/nar/gkm391 URL |
| [25] |
Sun L, Luo H, Bu D, et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts[J]. Nucleic Acids Res, 2013, 41(17):e166.
doi: 10.1093/nar/gkt646 URL |
| [26] |
Li A, Zhang J, Zhou Z. PLEK:a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme[J]. BMC Bioinformatics, 2014, 15:311.
doi: 10.1186/1471-2105-15-311 URL |
| [27] |
Finn RD, Coggill P, Eberhardt RY, et al. The Pfam protein families database:towards a more sustainable future[J]. Nucleic Acids Res, 2016, 44(d1):D279-D285.
doi: 10.1093/nar/gkv1344 URL |
| [28] |
Harrow J, Frankish A, Gonzalez JM, et al. GENCODE:the reference human genome annotation for The ENCODE Project[J]. Genome Res, 2012, 22(9):1760-1774.
doi: 10.1101/gr.135350.111 URL |
| [29] |
Chao Y, Yuan J, Guo T, et al. Analysis of transcripts and splice isoforms in Medicago sativa L. by single-molecule long-read sequencing[J]. Plant Mol Biol, 2019, 99(3):219-235.
doi: 10.1007/s11103-018-0813-y URL |
| [30] |
Wang T, Wang H, Cai D, et al. Comprehensive profiling of rhizome-associated alternative splicing and alternative polyadenylation in moso bamboo(Phyllostachys edulis)[J]. Plant J, 2017, 91(4):684-699.
doi: 10.1111/tpj.2017.91.issue-4 URL |
| [31] |
Ma JE, Jiang HY, Li LM, et al. SMRT sequencing of the full-length transcriptome of the Sunda pangolin(Manis javanica)[J]. Gene, 2019, 692:208-216.
doi: 10.1016/j.gene.2019.01.008 URL |
| [32] |
Li Y, Dai C, Hu C, et al. Global identification of alternative splicing via comparative analysis of SMRT-and Illumina-based RNA-seq in strawberry[J]. Plant J, 2017, 90(1):164-176.
doi: 10.1111/tpj.2017.90.issue-1 URL |
| [33] |
Liu X, Mei W, Soltis PS, et al. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome[J]. Mol Ecol Resour, 2017, 17(6):1243-1256.
doi: 10.1111/men.2017.17.issue-6 URL |
| [34] |
Bouba I, Kang Q, Luan YS, et al. Predicting miRNA-lncRNA interactions and recognizing their regulatory roles in stress response of plants[J]. Math Biosci, 2019, 312:67-76.
doi: 10.1016/j.mbs.2019.04.006 URL |
| [35] | 孙薏雯, 李金宝, 杨丹丹, 等. 长链非编码RNA在植物中的研究进展[J]. 山东农业大学学报:自然科学版, 2020, 51(5):968-974. |
| Sun YW, Li JB, Yang DD, et al. The research progress of long noncoding RNA in plants[J]. J Shandong Agric Univ:Nat Sci Ed, 2020, 51(5):968-974. | |
| [36] |
张楠, 刘自广, 孙世臣, 等. 拟南芥AtR8 lncRNA对盐胁迫响应及其对种子萌发的调节作用[J]. 植物学报, 2020, 55(4):421-429.
doi: 10.11983/CBB19244 |
| Zhang N, Liu ZG, Sun SC, et al. Response of AtR8 lncRNA to salt stress and its regulation on seed germination in Arabidopsis[J]. Chin Bull Bot, 2020, 55(4):421-429. |
| [1] | 李舒文, 李殷睿智, 董笛, 王梦迪, 晁跃辉, 韩烈保. 蒺藜苜蓿MtSAG113基因的转化及表达特征分析[J]. 生物技术通报, 2022, 38(1): 108-114. |
| [2] | 张业猛, 沈迎芳, 王英芳, 姚品雅, 王海庆. 蒺藜苜蓿MtLEA5B的克隆和功能分析[J]. 生物技术通报, 2018, 34(7): 101-107. |
| [3] | William C.Skarnes. 诱陷载体——一种哺乳类遗传学研究的新工具[J]. , 1991, 0(06): 1-5. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||