







生物技术通报 ›› 2021, Vol. 37 ›› Issue (8): 307-318.doi: 10.13560/j.cnki.biotech.bull.1985.2021-0183
王智博1,2( ), 王道平2, 苗兰1,3, 李瑛1, 潘映红2(), 刘建勋1,3()
), 王道平2, 苗兰1,3, 李瑛1, 潘映红2(), 刘建勋1,3()
收稿日期:2021-02-09
出版日期:2021-08-26
发布日期:2021-09-10
作者简介:王智博,男,硕士研究生,研究方向:中医药蛋白质组学;E-mail: 基金资助:
WANG Zhi-bo1,2(), WANG Dao-ping2, MIAO Lan1,3, LI Ying1, PAN Ying-hong2(), LIU Jian-xun1,3()
Received:2021-02-09
Published:2021-08-26
Online:2021-09-10
摘要:
比较和优化血液样本蛋白质组学样品制备和质谱分析技术,为深度研究和挖掘血液样本蛋白质组学信息创造条件。采用Q-Exactive Plus质谱仪,对比分析血浆、血清和去除高丰度蛋白血清预处理方法制备的大鼠血样蛋白质组构成;比较血清样本的常规酶切、45℃孵育、热辅助酶切、二次热辅助酶切、尿素辅助酶切和变温酶切的效率;比较数据依赖性采集(data-dependent acquisition,DDA)、数据非依赖性采集(data independent acquisition,DIA)和平行反应监测(parallel reaction monitoring,PRM)质谱数据采集的定性定量特征;采用优化的方法进行大鼠血液样本蛋白质组分析。血清样本去除高丰度蛋白后蛋白鉴定数更高、定量重复性更好;热辅助酶切和变温酶切血清样品的蛋白和肽段鉴定数以及质谱谱图匹配率相对较高,蛋白酶切效率和定性定量重复性较好;DDA操作简便,DIA重复性高,PRM定量精确;血清样本去除高丰度蛋白,采用热辅助结合变温酶切和DDA数据采集模式,3次重复试验分别鉴定到490、490、504个蛋白,鉴定总蛋白数590个,共有蛋白占比69.8%。优化的方法操作简单,蛋白鉴定率较高,重复性好,适用于血液样本的蛋白质组学分析。
王智博, 王道平, 苗兰, 李瑛, 潘映红, 刘建勋. 血液样本蛋白质组分析方法的比较研究[J]. 生物技术通报, 2021, 37(8): 307-318.
WANG Zhi-bo, WANG Dao-ping, MIAO Lan, LI Ying, PAN Ying-hong, LIU Jian-xun. Comparative Study on Methods of Analyzing Proteome in Blood Samples[J]. Biotechnology Bulletin, 2021, 37(8): 307-318.
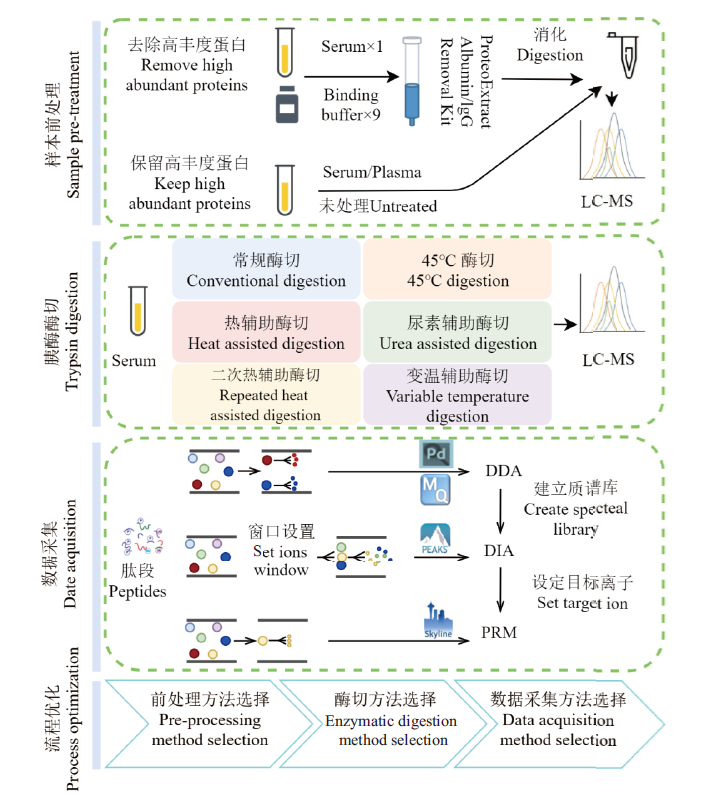
图1 实验流程
Fig.1 Experimental flow
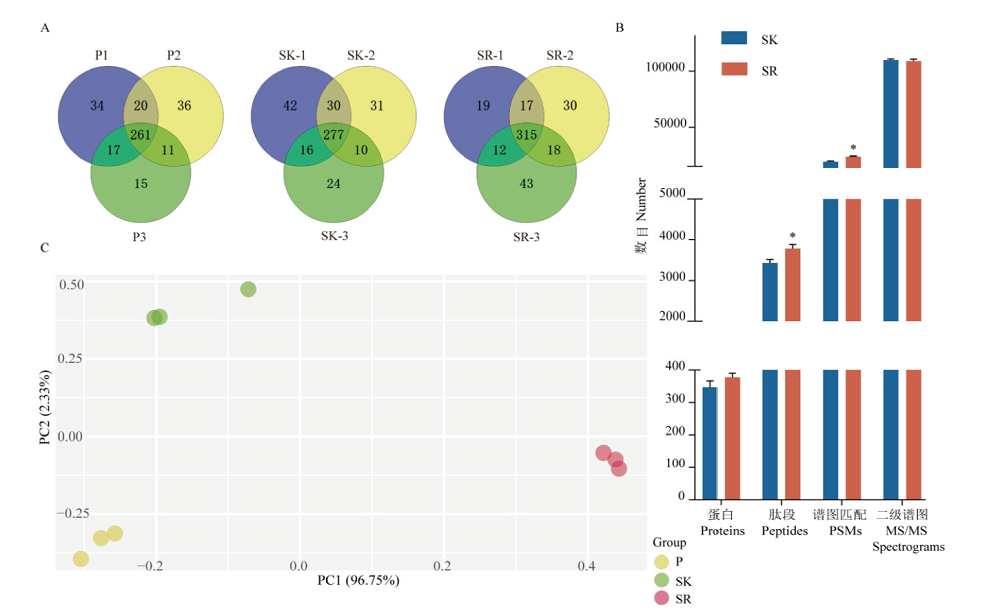
图2 三种预处理方法制备血样的质谱结果比较 A:血浆(P)、保留高丰度蛋白血清(SK)和去除高丰度蛋白血清(SR)三次重复制样蛋白鉴定数和重复率;B:SK和SR质谱鉴定蛋白数、肽段数、肽谱匹配数和二级谱图数的均值,**:P<0.01;C:蛋白定量主成分分析
Fig. 2 Comparison of mass spectrometry results of blood samples prepared by 3 pre-processing methods A:Protein identification numbers and duplication rates of three repeated sample preparations for plasma(P),serum(SK)with high-abundance proteins remained,and serum(SR)with high-abundance proteins removed. B:The average number of proteins,peptides,PSMs and MS/MS spectrum for SK and SR. **:P<0.01. C:Principal component analysis of protein quantification

图3 血清蛋白不同酶切方法质谱结果比较 A:鉴定蛋白数、肽段数、谱图匹配数的均值,*:P<0.05,**:P<0.01;B:蛋白遗漏酶切位点百分率比较;C:3次重复制样蛋白鉴定数和重复率;D:蛋白序列覆盖度;E:蛋白定量主成分分析
Fig. 3 Comparison of mass spectrometry results of serum proteins with different enzymatic digestion methods A:Average number of identified proteins,peptides and PSMs. *:P<0.05,and **:P<0.01. B:Comparison of percentage of missed enzymatic cleavage sites. C:Protein identification number and duplication rate of 3 repeated sample preparations. D:Sequence coverage of protein. E:Principal component analysis of protein quantification
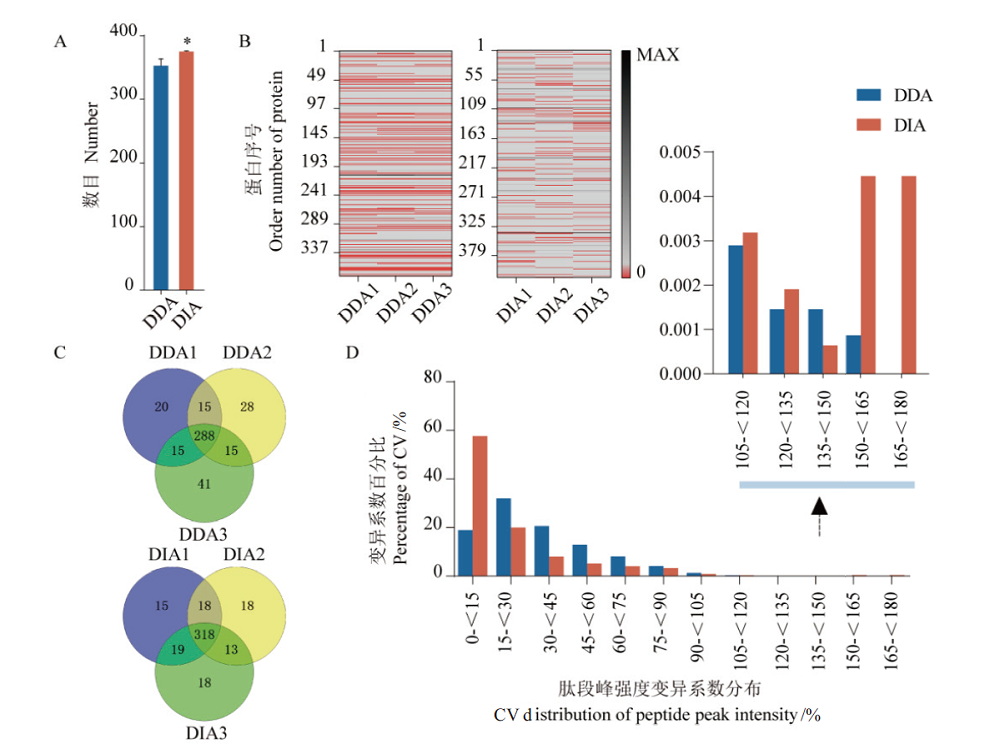
图4 DDA和DIA数据指标比较 A:3次技术重复蛋白鉴定数平均值,*P<0.05;B:3次技术重复蛋白定量缺失值矩阵,红色区域代表缺失值;C:3次技术重复蛋白定性重复性比较;D:肽段或蛋白峰强度变异系数分布统计
Fig. 4 Comparison of DDA and DIA data indices A:Average number of identified proteins in 3 technical repeats,*:P<0.05. B:Missing value matrix of protein quantification in 3 technical repeats,the red area represents missing values. C:Comparison of protein qualitative repeatability by 3 technical repeats. D:The statistics in coefficient of variation of the protein(peptides)peak intensity
| 蛋白 Protein | DIA* | PRM** |
|---|---|---|
| A0A0G2JSK1 | 0.163 | IFSQQADLSR,0.045;KIFSQQADLSR,0.064;DTLPHEDQGKGR,0.123 |
| A0A0G2JVP4 | 0.030 | ESATVTCLVK,0.058;TFPTLR,0.0732;DLPSPQK,0.128 |
| A0A0G2JV1 | 0.083 | HMEASLQEFKASPR,0.077;AVYLPNCDR,0.090;ISELKAEAVK,0.520 |
| A0A0G2K4K2 | 0.867 | LWIYDTSK,0.502 |
| A0A0G2K531 | 0.209 | QAALGAR,0.029;LFWEPMKIHDIR,0.460;NSCPPTAELLGSPGR,0.480 |
| A0A0G2K7X7 | 0.283 | CVGSAFETQSCNPER,0.019;ILPLTICK,0.019;ACGACPIWSK,0.032 |
| A0A0G2K896 | 0.217 | SSTFQLFGSPHGK,0.013;WCIVSDHEATK,0.059;VKWCAVGQQER,0.063 |
| B0BNN4 | 0.157 | SISCDEIPGQQSR,0.023;LCTPLLPK,0.031;HCYDIHNCVLK,0.129 |
| B2RYM3 | 0.052 | ELAAQTIK,0.024;ANLSSQILK,0.031;IADHKLSTFKADVR,0.049 |
| B5DEH7 | 0.114 | ANPGNFPWQAFTNIHGR,0.053;LPIADR,0.122;GLTVHLK,0.248 |
| D3ZAB3 | 0.711 | LMIYGATNLEDGVPSR,0.140 |
| D3ZAE6 | 1.732 | YLQGNTVQLR,0.085;QLDEGLFGR,0.450 |
| F1LWD0 | 0.867 | ANSYTTEYNPSVK,0.033 |
| F1LWS4 | 1.292 | NGYLYHENIRR,0.227;SYFPVPIGK,0.333;QCVFHYVENGESSYWQR,0.375 |
| F1LWW1 | 0.902 | VTISCR,1.485 |
| F1LXY6 | 0.040 | APEWLGFIR,0.037;ANGYTTEYNPSVK,0.040;AEDTATYYCAR,0.062 |
| F1LYU4 | 1.732 | ASNLASGIPAR,0.020 |
| G3V7L3 | 0.193 | TNVIQLR,0.019;CGTYGIYTK,0.066;LPITSLEK,0.080 |
| G3V7N9 | 0.055 | VITNVNDNYEPR,0.022;TVNSALRPNQAIR,0.057;TVNSALRPNQAIRFEK,0.146 |
| G3V7P2 | 0.250 | TLQEAVDSLKK,0.296 |
| G3V7P5 | 1.076 | WQSLPR,0.011;SCDVPVFENAK,0.022;IDHGSIKLPR,0.026 |
表1 DIA蛋白和对应肽段PRM 3次重复定量CV值分析
Table 1 Quantitative analysis on the coefficient of variation of protein from DIA and corresponding peptides from PRM in 3 repeats
| 蛋白 Protein | DIA* | PRM** |
|---|---|---|
| A0A0G2JSK1 | 0.163 | IFSQQADLSR,0.045;KIFSQQADLSR,0.064;DTLPHEDQGKGR,0.123 |
| A0A0G2JVP4 | 0.030 | ESATVTCLVK,0.058;TFPTLR,0.0732;DLPSPQK,0.128 |
| A0A0G2JV1 | 0.083 | HMEASLQEFKASPR,0.077;AVYLPNCDR,0.090;ISELKAEAVK,0.520 |
| A0A0G2K4K2 | 0.867 | LWIYDTSK,0.502 |
| A0A0G2K531 | 0.209 | QAALGAR,0.029;LFWEPMKIHDIR,0.460;NSCPPTAELLGSPGR,0.480 |
| A0A0G2K7X7 | 0.283 | CVGSAFETQSCNPER,0.019;ILPLTICK,0.019;ACGACPIWSK,0.032 |
| A0A0G2K896 | 0.217 | SSTFQLFGSPHGK,0.013;WCIVSDHEATK,0.059;VKWCAVGQQER,0.063 |
| B0BNN4 | 0.157 | SISCDEIPGQQSR,0.023;LCTPLLPK,0.031;HCYDIHNCVLK,0.129 |
| B2RYM3 | 0.052 | ELAAQTIK,0.024;ANLSSQILK,0.031;IADHKLSTFKADVR,0.049 |
| B5DEH7 | 0.114 | ANPGNFPWQAFTNIHGR,0.053;LPIADR,0.122;GLTVHLK,0.248 |
| D3ZAB3 | 0.711 | LMIYGATNLEDGVPSR,0.140 |
| D3ZAE6 | 1.732 | YLQGNTVQLR,0.085;QLDEGLFGR,0.450 |
| F1LWD0 | 0.867 | ANSYTTEYNPSVK,0.033 |
| F1LWS4 | 1.292 | NGYLYHENIRR,0.227;SYFPVPIGK,0.333;QCVFHYVENGESSYWQR,0.375 |
| F1LWW1 | 0.902 | VTISCR,1.485 |
| F1LXY6 | 0.040 | APEWLGFIR,0.037;ANGYTTEYNPSVK,0.040;AEDTATYYCAR,0.062 |
| F1LYU4 | 1.732 | ASNLASGIPAR,0.020 |
| G3V7L3 | 0.193 | TNVIQLR,0.019;CGTYGIYTK,0.066;LPITSLEK,0.080 |
| G3V7N9 | 0.055 | VITNVNDNYEPR,0.022;TVNSALRPNQAIR,0.057;TVNSALRPNQAIRFEK,0.146 |
| G3V7P2 | 0.250 | TLQEAVDSLKK,0.296 |
| G3V7P5 | 1.076 | WQSLPR,0.011;SCDVPVFENAK,0.022;IDHGSIKLPR,0.026 |
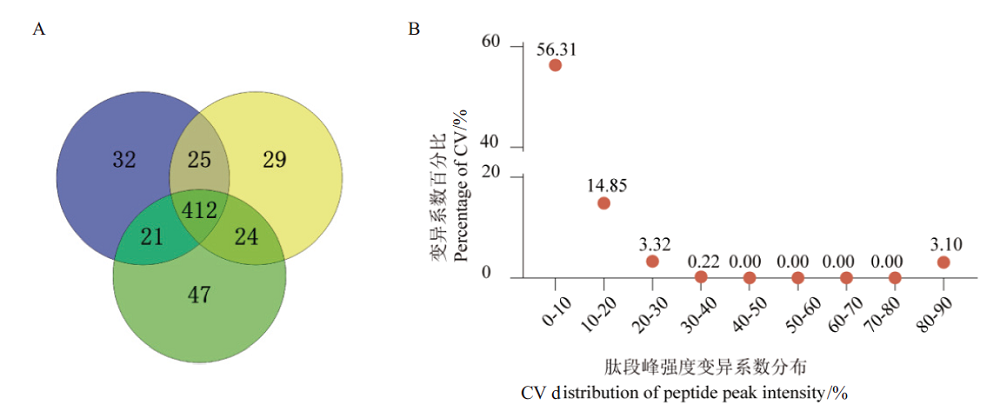
图5 基于优化流程分析的血样蛋白组定性和定量结果 A:DDA定性结果Venn图;B:DDA蛋白定量CV值分布图
Fig.5 Qualitative and quantitative results of blood sample proteome based on optimized workflow A:Venn diagram of DDA qualitative results. B:CV distribution diagram of DDA protein quantitative results
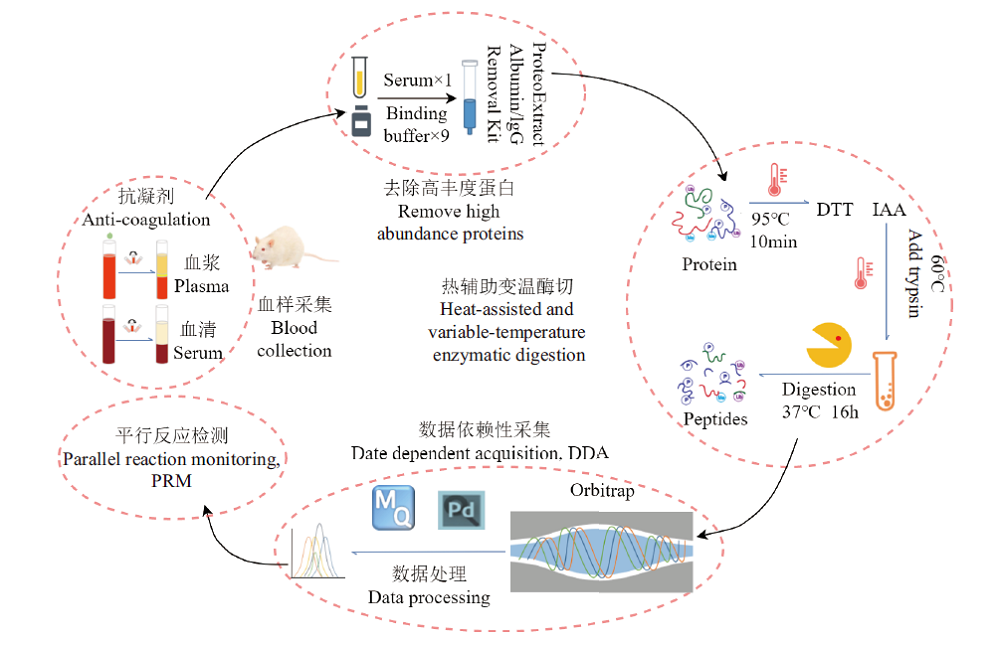
图6 血样蛋白质组样品制备和分析流程
Fig. 6 Sample preparation and analysis workflow for blood proteome
| [1] |
Aslam B, Basit M, Nisar MA, et al. Proteomics:technologies and their applications[J]. J Chromatogr Sci, 2017, 55(2):182-196.
doi: 10.1093/chromsci/bmw167 URL |
| [2] |
Geyer PE, Holdt LM, Teupser D, et al. Revisiting biomarker discovery by plasma proteomics[J]. Mol Syst Biol, 2017, 13(9):942.
doi: 10.15252/msb.20156297 URL |
| [3] | Geyer PE, Kulak NA, Pichler G, et al. Plasma proteome profiling to assess human health and disease[J]. Cell Syst, 2016, 2(3):185-195. |
| [4] |
Peng L, Cantor DI, Huang C, et al. Tissue and plasma proteomics for early stage cancer detection[J]. Mol Omics, 2018, 14(6):405-423.
doi: 10.1039/C8MO00126J URL |
| [5] | 牟永莹, 王道平, 陈明, 等. 大豆种子蛋白质组样品制备与数据分析方法[J]. 生物技术通报, 2020, 36(12):247-255. |
| Mu YY, Wang DP, Chen M, et al. Sample preparation and data analysis method for soybean seed proteome[J]. Biotechnol Bull, 2020, 36(12):247-255. | |
| [6] | Wiśniewski JR. Filter-aided sample preparation:the versatile and efficient method for proteomic analysis[J]. Methods Enzymol, 2017, 585:15-27. |
| [7] |
Zolotarjova N, Martosella J, Nicol G, et al. Differences among techniques for high-abundant protein depletion[J]. Proteomics, 2005, 5(13):3304-3313.
pmid: 16052628 |
| [8] |
Polaskova V, Kapur A, Khan A, et al. High-abundance protein depletion:comparison of methods for human plasma biomarker discovery[J]. Electrophoresis, 2010, 31(3):471-482.
doi: 10.1002/elps.200900286 pmid: 20119956 |
| [9] |
Lima-Oliveira G, Monneret D, Guerber F, et al. Sample management for clinical biochemistry assays:Are serum and plasma interchangeable specimens?[J]. Crit Rev Clin Lab Sci, 2018, 55(7):480-500.
doi: 10.1080/10408363.2018.1499708 URL |
| [10] |
Park ZY, Russell DH. Thermal denaturation:a useful technique in peptide mass mapping[J]. Anal Chem, 2000, 72(11):2667-2670.
pmid: 10857653 |
| [11] |
Schniers A, Pasing Y, Hansen T. Lys-C/trypsin tandem-digestion protocol for gel-free proteomic analysis of colon biopsies[J]. Methods Mol Biol, 2019, 1959:113-122.
doi: 10.1007/978-1-4939-9164-8_7 pmid: 30852818 |
| [12] |
Betancourt LH, Sanchez A, Pla I, et al. Quantitative assessment of urea in-solution Lys-C/trypsin digestions reveals superior performance at room temperature over traditional proteolysis at 37℃[J]. J Proteome Res, 2018, 17(7):2556-2561.
doi: 10.1021/acs.jproteome.8b00228 pmid: 29812944 |
| [13] |
Budnik B, Levy E, Harmange G, et al. SCoPE-MS:mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation[J]. Genome Biol, 2018, 19(1):161.
doi: 10.1186/s13059-018-1547-5 URL |
| [14] |
Hu A, Noble WS, Wolf-Yadlin A. Technical advances in proteomics:new developments in data-independent acquisition[J]. F1000 Research, 2016, 5:419.
doi: 10.12688/f1000research URL |
| [15] |
Zhang Y, Fonslow BR, Shan B, et al. Protein analysis by shotgun/bottom-up proteomics[J]. Chem Rev, 2013, 113(4):2343-2394.
doi: 10.1021/cr3003533 URL |
| [16] |
Barkovits K, Pacharra S, Pfeiffer K, et al. Reproducibility, specificity and accuracy of relative quantification using spectral library-based data-independent acquisition[J]. Mol Cell Proteomics, 2020, 19(1):181-197.
doi: 10.1074/mcp.RA119.001714 pmid: 31699904 |
| [17] |
Venable JD, Dong MQ, Wohlschlegel J, et al. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra[J]. Nat Methods, 2004, 1(1):39-45.
pmid: 15782151 |
| [18] |
Bilbao A, Varesio E, Luban J, et al. Processing strategies and software solutions for data-independent acquisition in mass spectrometry[J]. Proteomics, 2015, 15(5/6):964-980.
doi: 10.1002/pmic.201400323 URL |
| [19] |
Shi T, Song E, Nie S, et al. Advances in targeted proteomics and applications to biomedical research[J]. Proteomics, 2016, 16(15/16):2160-2182.
doi: 10.1002/pmic.v16.15-16 URL |
| [20] |
Uzozie AC, Aebersold R. Advancing translational research and precision medicine with targeted proteomics[J]. J Proteomics, 2018, 189:1-10.
doi: 10.1016/j.jprot.2018.02.021 URL |
| [21] |
Silva ARM, Toyoshima MTK, Passarelli M, et al. Comparing data-independent acquisition and parallel reaction monitoring in their abilities to differentiate high-density lipoprotein subclasses[J]. J Proteome Res, 2020, 19(1):248-259.
doi: 10.1021/acs.jproteome.9b00511 URL |
| [22] |
Chiva C, Ortega M, Sabidó E. Influence of the digestion technique, protease, and missed cleavage peptides in protein quantitation[J]. J Proteome Res, 2014, 13(9):3979-3986.
doi: 10.1021/pr500294d URL |
| [23] |
Bruderer R, Muntel J, Müller S, et al. Analysis of 1508 plasma samples by capillary-flow data-independent acquisition profiles proteomics of weight loss and maintenance[J]. Mol Cell Proteomics, 2019, 18(6):1242-1254.
doi: 10.1074/mcp.RA118.001288 pmid: 30948622 |
| [24] |
Manes NP, Nita-Lazar A. Application of targeted mass spectrometry in bottom-up proteomics for systems biology research[J]. J Proteom, 2018, 189:75-90.
doi: 10.1016/j.jprot.2018.02.008 URL |
| [25] |
Woo CM, Lund PJ, Huang AC, et al. Mapping and quantification of over 2000 O-linked glycopeptides in activated human T cells with isotope-targeted glycoproteomics(isotag)[J]. Mol Cell Proteom, 2018, 17(4):764-775.
doi: 10.1074/mcp.RA117.000261 URL |
| [26] |
Qiu FH, Hou TY, Huang DH, et al. Evaluation of two high-abundance protein depletion kits and optimization of downstream isoelectric focusing[J]. Mol Med Rep, 2015, 12(5):7749-7755.
doi: 10.3892/mmr.2015.4417 URL |
| [27] |
Liu B, Qiu FH, Voss C, et al. Evaluation of three high abundance protein depletion kits for umbilical cord serum proteomics[J]. Proteome Sci, 2011, 9(1):24.
doi: 10.1186/1477-5956-9-24 URL |
| [28] |
Glatter T, Ludwig C, Ahrné E, et al. Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys-C/trypsin proteolysis over trypsin digestion[J]. J Proteome Res, 2012, 11(11):5145-5156.
doi: 10.1021/pr300273g pmid: 23017020 |
| [29] |
Kailasa S, Wu HF. Advances in nanomaterial-based microwaves and infrared wave-assisted tryptic digestion for ultrafast proteolysis and rapid detection by MALDI-MS[J]. Comb Chem High Throughput Screen, 2014, 17(1):68-79.
doi: 10.2174/1386207316666131110211353 URL |
| [1] | 徐美慧, 张耀杰, 唐克轩, 苗志奇. GoldenBraid酶切连接反应体系的优化[J]. 生物技术通报, 2020, 36(9): 266-274. |
| [2] | 孟丽娜, 彭春莹, 李铁栋, 李博生. 基于蛋白质组学对螺旋藻砷胁迫响应机制的研究[J]. 生物技术通报, 2020, 36(4): 107-116. |
| [3] | 李堃, 刘悦, 黄鹏, 杨智昉, 胡茜, 张颖, 李志宏, 吕叶辉, 梁乐. 小鼠精原细胞分化的蛋白质组学研究[J]. 生物技术通报, 2020, 36(3): 168-176. |
| [4] | 牟永莹, 王道平, 陈明, 邱丽娟, 潘映红. 大豆种子蛋白质组样品制备与数据分析方法[J]. 生物技术通报, 2020, 36(12): 247-255. |
| [5] | 林美璇, 周小满, 关锋, 崔文璟. 磷脂酰肌醇特异性磷脂酶C的异源表达和应用[J]. 生物技术通报, 2020, 36(1): 81-87. |
| [6] | 张良, 陈小青, 宋佳宇, 毛然然, 姜倩雯, 林向民. 巴洛沙星胁迫下大肠杆菌的比较蛋白质组学研究[J]. 生物技术通报, 2019, 35(3): 103-109. |
| [7] | 兰玉婷, 王双蕾, 李征珍, 冯金朝, 王晓东, 石莎. 沙冬青属植物响应非生物胁迫的蛋白质组学研究进展[J]. 生物技术通报, 2019, 35(1): 112-119. |
| [8] | 牟永莹,顾培明,马博,闫文秀,王道平,潘映红. 基于质谱的定量蛋白质组学技术发展现状[J]. 生物技术通报, 2017, 33(9): 73-84. |
| [9] | 邵贵芳, 张凡, 王姣, 赵凯, 莫云容, 邓明华. 辣椒雄性不育的研究进展[J]. 生物技术通报, 2017, 33(8): 7-13. |
| [10] | 余乐正, 柳凤娟, 吴正雨, 冉小强. 分泌蛋白质组学在肿瘤标志物中的研究进展[J]. 生物技术通报, 2017, 33(3): 12-21. |
| [11] | 孔德康, 王红旗, 许洁, 刘自力, 吴枭雄. 基因组学、蛋白质组学和代谢组学在微生物降解PAHs中的应用[J]. 生物技术通报, 2017, 33(10): 46-51. |
| [12] | 卢曾奎,马友记,. 定量蛋白质组学在动物睾丸蛋白研究中的应用进展[J]. 生物技术通报, 2016, 32(12): 8-12. |
| [13] | 马亚龙,刘红昌,夏金兰,杨云,聂珍媛,. 极端嗜酸热古菌Acidianus manzaensis胞外硫活化蛋白质基因的筛选及鉴定[J]. 生物技术通报, 2016, 32(12): 143-151. |
| [14] | 冯毛,李建科. 蜜蜂蛋白质组学研究新进展[J]. 生物技术通报, 2015, 31(4): 92-98. |
| [15] | 崔学强,张树珍,沈林波,冯翠莲. 转基因甘蔗植株Southern杂交体系的优化[J]. 生物技术通报, 2015, 31(12): 105-109. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||