







生物技术通报 ›› 2025, Vol. 41 ›› Issue (11): 301-310.doi: 10.13560/j.cnki.biotech.bull.1985.2025-0366
• 研究报告 • 上一篇
蒋天威1( ), 李亚娇2, 马培杰2, 陈才俊2, 刘晓霞2, 陈莹2(), 王小利2()
), 李亚娇2, 马培杰2, 陈才俊2, 刘晓霞2, 陈莹2(), 王小利2()
收稿日期:2025-04-07
出版日期:2025-11-26
发布日期:2025-12-09
通讯作者:
王小利,男,研究员,研究方向 :分子植物学;E-mail: WangXiaoli_GIP@163.com作者简介:蒋天威,男,硕士研究生,研究方向 :分子植物育种;E-mail: JiangTianwei_GIP@163.com
基金资助:
JIANG Tian-wei1(), LI Ya-jiao2, MA Pei-jie2, CHEN Cai-jun2, LIU Xiao-xia2, CHEN Ying2(), WANG Xiao-li2()
Received:2025-04-07
Published:2025-11-26
Online:2025-12-09
摘要:
目的 探索在长日照条件下,蒺藜苜蓿开花过程中的DNA甲基化变化,探究光周期相关基因与DNA甲基化的可能关系。 方法 利用全基因组亚硫酸氢盐测序(WGBS)检测营养期和开花期的蒺藜苜蓿叶片,分析DNA甲基化差异。 结果 (1)甲基化类型主要为CG型(69.74%),其次是CHG(35.22%)和CHH(21.81%);基因上游的CHG甲基化,以及基因上下游及基因体区域的CHH甲基化水平均是营养期比开花期的高。(2)差异甲基化区域(DMR)共有20 647个,其中CHH型最多(11 247个),且68%为低甲基化区域。(3)基于基因本体(GO)和信号通路(KEGG)对全部DMR关联基因(DMG)进行分析,发现DMG主要参与在高亲和力寡肽跨膜转运蛋白活性、FAD结合核苷三磷酸酶活性、ATP酶活性、水解酶活性,作用于酸酐四氢叶酸生物合成过程等通路中。(4)参与光周期开花途径关键基因CRY1、CRY2、FKF1、PHYA、ELF3、COL2、FT、LHY、ZTL都发生了显著的甲基化水平改变。 结论 DNA甲基化可能参与调控蒺藜苜蓿开花,探索了蒺藜苜蓿光周期诱导成花过程中DNA甲基化的可能角色,为蒺藜苜蓿育种研究提供新见解。
蒋天威, 李亚娇, 马培杰, 陈才俊, 刘晓霞, 陈莹, 王小利. 蒺藜苜蓿开花过程中的全基因组DNA甲基化分析[J]. 生物技术通报, 2025, 41(11): 301-310.
JIANG Tian-wei, LI Ya-jiao, MA Pei-jie, CHEN Cai-jun, LIU Xiao-xia, CHEN Ying, WANG Xiao-li. Whole-genome DNA Methylation Analysis during the Flowering Processof Medicago truncatula[J]. Biotechnology Bulletin, 2025, 41(11): 301-310.
样本 Sample | 总读取数 Total reads | 比对读取数 Mapped reads | 唯一比对读取数 Uniquely reads | 测序深度 Seqencing depth | 唯一比对率 Uniquely mapped reads (%) | 比对率 Mapping rate (%) | Q30 (%) | 转换率 Conversion rate (%) |
|---|---|---|---|---|---|---|---|---|
| FP_1 | 107573870 | 62923962 | 43437098 | 34.38 | 40.38 | 58.49 | 90.98 | 99.69 |
| FP_2 | 93350734 | 52038408 | 34920690 | 29.86 | 37.41 | 55.75 | 91.34 | 99.67 |
| FP_3 | 114432912 | 66064890 | 45284390 | 36.65 | 39.57 | 57.73 | 91.97 | 99.65 |
| NP_1 | 103921110 | 60296822 | 44356076 | 33.25 | 42.68 | 58.02 | 91.44 | 99.67 |
| NP_2 | 120328788 | 72294208 | 50808854 | 38.52 | 42.23 | 60.08 | 91.87 | 99.59 |
| NP_3 | 129107620 | 74594082 | 51931090 | 41.32 | 40.22 | 57.78 | 91.59 | 99.61 |
表1 蒺藜苜蓿全基因组甲基化测序数据处理
Table 1 Processing of whole-genome methylation sequencing data of Medicago truncatula
样本 Sample | 总读取数 Total reads | 比对读取数 Mapped reads | 唯一比对读取数 Uniquely reads | 测序深度 Seqencing depth | 唯一比对率 Uniquely mapped reads (%) | 比对率 Mapping rate (%) | Q30 (%) | 转换率 Conversion rate (%) |
|---|---|---|---|---|---|---|---|---|
| FP_1 | 107573870 | 62923962 | 43437098 | 34.38 | 40.38 | 58.49 | 90.98 | 99.69 |
| FP_2 | 93350734 | 52038408 | 34920690 | 29.86 | 37.41 | 55.75 | 91.34 | 99.67 |
| FP_3 | 114432912 | 66064890 | 45284390 | 36.65 | 39.57 | 57.73 | 91.97 | 99.65 |
| NP_1 | 103921110 | 60296822 | 44356076 | 33.25 | 42.68 | 58.02 | 91.44 | 99.67 |
| NP_2 | 120328788 | 72294208 | 50808854 | 38.52 | 42.23 | 60.08 | 91.87 | 99.59 |
| NP_3 | 129107620 | 74594082 | 51931090 | 41.32 | 40.22 | 57.78 | 91.59 | 99.61 |

图1 蒺藜苜蓿从营养期到开花期DNA甲基化的变化A:样品相关性分析;B:3种序列下甲基化水平统计;C:基因体区域及其侧翼区域内3种序列的甲基化的分布。FP为开花期,NP为营养期。下同
Fig. 1 Variations in DNA methylation from the vegetative stage to the flowering stage in M. truncatulaA: Sample correlation analysis. B: Methylation level statistics for the three sequences. C: Methylation distribution in gene body regions and their flanking regions for the three sequences. FP refers to the flowering stage, and NP refers to the vegetative stage. The same below
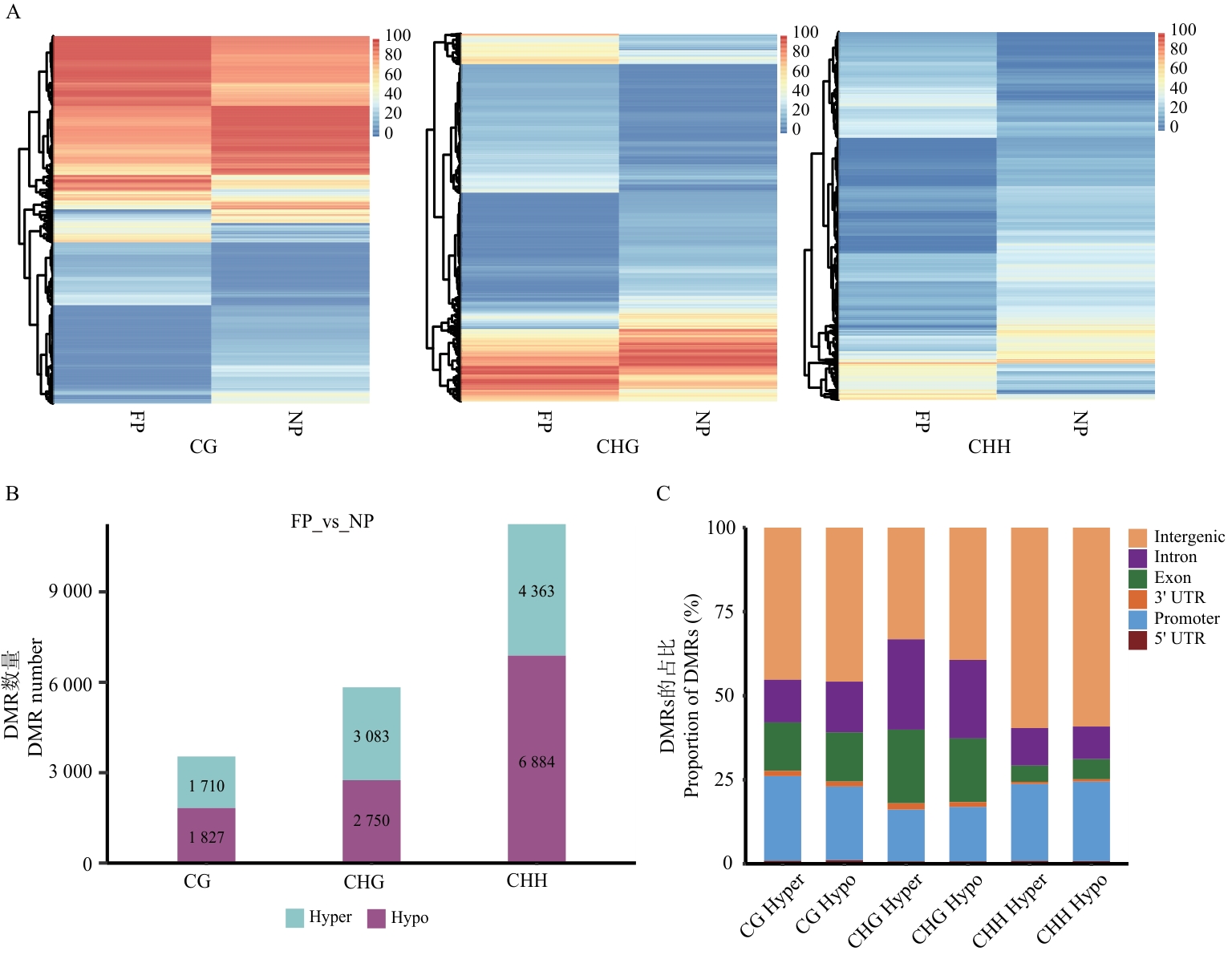
图2 FP vs NP中差异甲基化区域分析A:3种序列下差异甲基化区域聚类热图;B:差异甲基化区域数量统计图;C:差异甲基化区域在基因组的分布比率图。图B、C中Hyper为高甲基化,Hypo为低甲基化;图C中图例展示了基因间区(Intergenic)、内含子(Intron)、外显子(Exon)、3'非翻译区(3' UTR)、启动子(Promoter)和5'非翻译区(5' UTR)
Fig. 2 Differentially methylated region analysis between FP and NPA: Clustering heatmap of differentially methylated regions (DMRs) in the three sequences. B: Statistical chart of the number of DMRs. C: Proportional distribution of DMRs in the genome. In figure B and C, Hyper indicates hypermethylation, and Hypo indicates hypomethylation

图3 FP vs NP的CG 、CHG和CHH类型DMR 关联基因的 GO(A、B、C)、KEGG(D、E、F) 富集分析PPAH: hydrolase activity, acting on acid anhydrides, in phosphorus-containing anhydrides
Fig. 3 GO (A, B, C) and KEGG (D, E, F) enrichment analysis of genes associated with CG, CHG, and CHH types of DMRs in FP vs NP
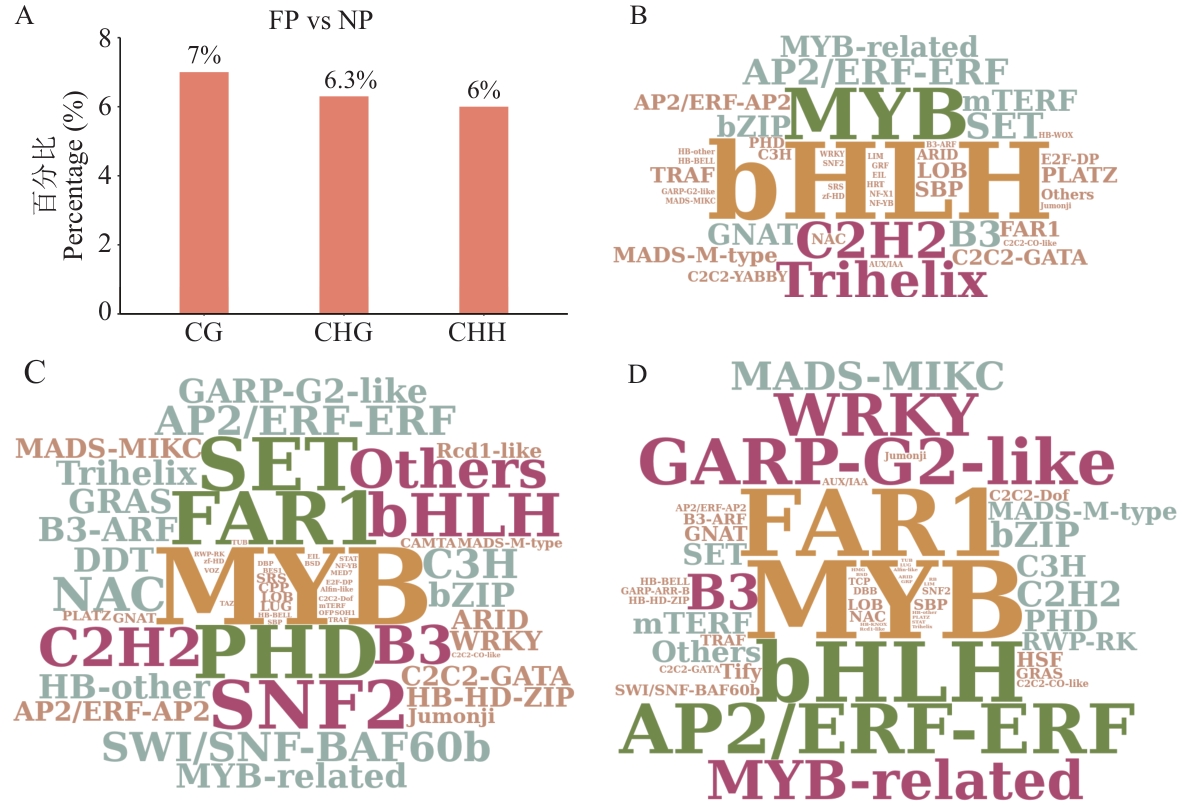
图4 DMRs关联的转录因子分析A: FP vs NP中DMR关联基因中转录因子编码基因的比例;B、C、D:3种序列背景下DMR关联转录因子家族丰度词云图。图B对应CG、图C对应CHG、图D对应CHH;图B、C、D中文字越大丰度越高
Fig. 4 Transcription factor analysis associated with DMRsA: Proportion of transcription factor-encoding genes among DMR-associated genes in FP vs NP. B, C, D: Word clouds showing the abundance of DMR-associated transcription factor families under three sequence contexts. Figure B corresponds to CG, Figure C to CHG, and Figure D to CHH; the larger the text in Figure B, C, and D, the higher the abundance

图5 转录因子编码基因的蛋白互作网络图A:CG序列下蛋白互作网络;B:CHG序列下蛋白互作网络;C:CHH序列下蛋白互作网络。节点代表蛋白,线代表互作关系,节点旁红色文字为关键节点基因编号
Fig. 5 Protein-protein interaction network of transcription factor-encoding genesA: Protein-protein interaction network under CG context. B: Protein-protein interaction network under CHG context. C: Protein-protein interaction network under CHH context. Nodes indicate proteins, edges indicate interaction relationships, and red text next to the nodes indicates the gene IDs of key nodes
序列类型 Type | 基因 Gene | 去甲基化 Hypo-DMR | 超甲基化 Hyper-DMR |
|---|---|---|---|
| CG | ACTR4 | - | Up |
| GA20ox1 | - | Up | |
| CRY1 | - | Up | |
| UBOX13 | - | Up | |
| NFYB3 | - | Up | |
| CRY2 | - | Up | |
| MYB1 | - | Up | |
| CHS | - | Up | |
| SRO1 | Down | - | |
| BHLH130 | Down | - | |
| CHG | CCX5 | - | Up |
| VOZ1 | - | Up | |
| ACTR4 | - | Up | |
| ATX2 | - | Up | |
| FKF1 | - | Up | |
| MYB75 | - | Up | |
| PHYA | - | Up | |
| ELF3 | - | Up | |
| UPF2 | Down | - | |
| ELF6 | Down | - | |
| BRN1 | Down | - | |
| CHS | Down | - | |
| COL 2 | Down | - | |
| CHH | EZA1 | - | Up |
| PEPPER | - | Up | |
| MYB75 | - | Up | |
| CSNK2A | - | Up | |
| CHS9 | - | Up | |
| CHS2 | Down | - | |
| DMR6-LIKE OXYGENASE 2 | Down | - | |
| RENT3 | Down | - | |
| FD | Down | - | |
| CCA1 | Down | - | |
| DMR6-LIKE OXYGENASE 2 | Down | - | |
| bHLH122 | Down | - | |
| MYST family 2 | Down | - | |
| COL 9 | Down | - | |
| EZA1 | Down | - | |
| BRN1 | Down | - | |
| AGL18 | Down | - | |
| FT | Down | - | |
| MYB114 | Down | - | |
| LHY | Down | - | |
| MYB1 | Down | - | |
| CHS | Down | - | |
| ZTL | Down | - | |
| COL2 | Down | - |
表2 与光周期相关的DMR关联基因
Table 2 DMR-associated genes related to photoperiod
序列类型 Type | 基因 Gene | 去甲基化 Hypo-DMR | 超甲基化 Hyper-DMR |
|---|---|---|---|
| CG | ACTR4 | - | Up |
| GA20ox1 | - | Up | |
| CRY1 | - | Up | |
| UBOX13 | - | Up | |
| NFYB3 | - | Up | |
| CRY2 | - | Up | |
| MYB1 | - | Up | |
| CHS | - | Up | |
| SRO1 | Down | - | |
| BHLH130 | Down | - | |
| CHG | CCX5 | - | Up |
| VOZ1 | - | Up | |
| ACTR4 | - | Up | |
| ATX2 | - | Up | |
| FKF1 | - | Up | |
| MYB75 | - | Up | |
| PHYA | - | Up | |
| ELF3 | - | Up | |
| UPF2 | Down | - | |
| ELF6 | Down | - | |
| BRN1 | Down | - | |
| CHS | Down | - | |
| COL 2 | Down | - | |
| CHH | EZA1 | - | Up |
| PEPPER | - | Up | |
| MYB75 | - | Up | |
| CSNK2A | - | Up | |
| CHS9 | - | Up | |
| CHS2 | Down | - | |
| DMR6-LIKE OXYGENASE 2 | Down | - | |
| RENT3 | Down | - | |
| FD | Down | - | |
| CCA1 | Down | - | |
| DMR6-LIKE OXYGENASE 2 | Down | - | |
| bHLH122 | Down | - | |
| MYST family 2 | Down | - | |
| COL 9 | Down | - | |
| EZA1 | Down | - | |
| BRN1 | Down | - | |
| AGL18 | Down | - | |
| FT | Down | - | |
| MYB114 | Down | - | |
| LHY | Down | - | |
| MYB1 | Down | - | |
| CHS | Down | - | |
| ZTL | Down | - | |
| COL2 | Down | - |
| [23] | Taoka KI, Ohki I, Tsuji H, et al. 14-3-3 proteins act as intracellular receptors for rice Hd3a florigen [J]. Nature, 2011, 476(7360): 332-335. |
| [24] | Qin ZR, Bai YX, Muhammad S, et al. Divergent roles of FT-like 9 in flowering transition under different day lengths in Brachypodium distachyon [J]. Nat Commun, 2019, 10(1): 812. |
| [25] | Yang ZF, Yan HD, Nie G, et al. Induction of flowering under long-day photoperiod requires DNA hypermethylation in orchardgrass [J]. J Exp Bot, 2025, 76(9): 2557-2572. |
| [26] | Zhou Q, Jia CL, Ma WX, et al. MYB transcription factors in alfalfa (Medicago sativa): genome-wide identification and expression analysis under abiotic stresses [J]. PeerJ, 2019, 7: e7714. |
| [27] | Zhang LC, Liu GX, Jia JZ, et al. The wheat MYB-related transcription factor TaMYB72 promotes flowering in rice [J]. J Integr Plant Biol, 2016, 58(8): 701-704. |
| [28] | Zhu L, Guan YX, Liu YN, et al. Regulation of flowering time in Chrysanthemum by the R2R3 MYB transcription factor CmMYB2 is associated with changes in gibberellin metabolism [J]. Hortic Res, 2020, 7: 96. |
| [29] | Liu YW, Li X, Li KW, et al. Multiple bHLH proteins form heterodimers to mediate CRY2-dependent regulation of flowering-time in Arabidopsis [J]. PLoS Genet, 2013, 9(10): e1003861. |
| [30] | Aslam M, Jakada BH, Fakher B, et al. Genome-wide study of pineapple (Ananas comosus L.) bHLH transcription factors indicates that cryptochrome-interacting bHLH2 (AcCIB2) participates in flowering time regulation and abiotic stress response [J]. BMC Genomics, 2020, 21(1): 735. |
| [31] | Zhou Q, Cui Y, Dong R, et al. Integrative analyses of transcriptomes and metabolomes reveal associated genes and metabolites with flowering regulation in common vetch (Vicia sativa L.) [J]. Int J Mol Sci, 2022, 23(12): 6818. |
| [32] | Sun H, Guo ZA, Gao LF, et al. DNA methylation pattern of Photoperiod-B1 is associated with photoperiod insensitivity in wheat (Triticum aestivum) [J]. New Phytol, 2014, 204(3): 682-692. |
| [33] | Sun Q, Qiao J, Zhang S, et al. Changes in DNA methylation assessed by genomic bisulfite sequencing suggest a role for DNA methylation in cotton fruiting branch development [J]. PeerJ, 2018, 6: e4945. |
| [34] | Song QX, Zhang TZ, Stelly DM, et al. Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons [J]. Genome Biol, 2017, 18(1): 99. |
| [1] | Qi QY, Hu BC, Jiang WY, et al. Advances in plant epigenome editing research and its application in plants [J]. Int J Mol Sci, 2023, 24(4): 3442. |
| [2] | Qiao S, Song W, Hu WT, et al. The role of plant DNA methylation in development, stress response, and crop breeding [J]. Agronomy (Basel), 2024, 15(1): 94. |
| [3] | Zeng YB, Dawe RK, Gent JI. Natural methylation epialleles correlate with gene expression in maize [J]. Genetics, 2023, 225(2): iyad146. |
| [4] | Mahmood T, He SP, Abdullah M, et al. Epigenetic insight into floral transition and seed development in plants [J]. Plant Sci, 2024, 339: 111926. |
| [5] | Kumar S, Mohapatra T. Dynamics of DNA methylation and its functions in plant growth and development [J]. Front Plant Sci, 2021, 12: 596236. |
| [6] | Sun M. Advances in plant epigenetic regulation of abiotic stress response[J]. Theoretical and Natural Science, 2025, 90: 81-87. |
| [7] | Rehman S, Bahadur S, Xia W. An overview of floral regulatory genes in annual and perennial plants [J]. Gene, 2023, 885: 147699. |
| [8] | Shi ZY, Zhao WQ, Li CR, et al. Overexpression of the Chrysanthemum lavandulifolium ROS1 gene promotes flowering in Arabidopsis thaliana by reducing the methylation level of CONSTANS [J]. Plant Sci, 2024, 342: 112019. |
| [9] | Shi MM, Wang CL, Wang P, et al. Role of methylation in vernalization and photoperiod pathway: a potential flowering regulator? [J]. Hortic Res, 2023, 10(10): uhad174. |
| [10] | Xie HJ, Li XC, Sun YL, et al. DNA methylation of the autonomous pathway is associated with flowering time variations in Arabidopsis thaliana [J]. Int J Mol Sci, 2024, 25(13): 7478. |
| [11] | Sala-Cholewa K, Tomasiak A, Nowak K, et al. DNA methylation analysis of floral parts revealed dynamic changes during the development of homostylous Fagopyrum tataricum and heterostylous F. esculentum flowers [J]. BMC Plant Biol, 2024, 24(1): 448. |
| [12] | He GM, Zhu XP, Elling AA, et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids [J]. Plant Cell, 2010, 22(1): 17-33. |
| [35] | Hsieh TF, Ibarra CA, Silva P, et al. Genome-wide demethylation of Arabidopsis endosperm [J]. Science, 2009, 324(5933): 1451-1454. |
| [13] | Xu W, Yang TQ, Dong X, et al. Genomic DNA methylation analyses reveal the distinct profiles in Castor bean seeds with persistent endosperms [J]. Plant Physiol, 2016, 171(2): 1242-1258. |
| [14] | Vagner B, Senjuti S, Yun K, et al. The Medicago Truncatula Genome [M]. Cham, Switzerland : Springer, 2022. |
| [15] | Beck D, Ben Maamar M, Skinner MK. Genome-wide CpG density and DNA methylation analysis method (MeDIP, RRBS, and WGBS) comparisons [J]. Epigenetics, 2022, 17(5): 518-530. |
| [16] | Noh B, Lee SH, Kim HJ, et al. Divergent roles of a pair of homologous jumonji/zinc-finger-class transcription factor proteins in the regulation of Arabidopsis flowering time [J]. Plant Cell, 2004, 16(10): 2601-2613. |
| [17] | Lin XW, Yuan TT, Guo H, et al. The regulation of chromatin configuration at AGAMOUS locus by LFR-SYD-containing complex is critical for reproductive organ development in Arabidopsis [J]. Plant J, 2023, 116(2): 478-496. |
| [18] | Sachdev S, Biswas R, Roy A, et al. The Arabidopsis arid-hmg dna-binding protein 15 modulates jasmonic acid signaling by regulating MYC2 during pollen development [J]. Plant Physiol, 2024, 196(2): 996-1013. |
| [19] | Bartels A, Han Q, Nair P, et al. Dynamic DNA methylation in plant growth and development [J]. Int J Mol Sci, 2018, 19(7): 2144. |
| [20] | Kondo H, Shiraya T, Wada KC, et al. Induction of flowering by DNA demethylation in Perilla frutescens and Silene armeria: Heritability of 5-azacytidine-induced effects and alteration of the DNA methylation state by photoperiodic conditions [J]. Plant Sci, 2010, 178(3): 321-326. |
| [21] | Yang HX, Chang F, You CJ, et al. Whole-genome DNA methylation patterns and complex associations with gene structure and expression during flower development in Arabidopsis [J]. Plant J, 2015, 81(2): 268-281. |
| [22] | Wu X, Chen SY, Lin F, et al. Comparative and functional analysis unveils the contribution of photoperiod to DNA methylation, sRNA accumulation, and gene expression variations in short-day and long-day grasses [J]. Plant J, 2024, 118(6): 1955-1971. |
| [1] | 陈强, 于璎霏, 张颖, 张冲. 茉莉酸甲酯对薄皮甜瓜‘绿宝石’采后冷害的调控[J]. 生物技术通报, 2025, 41(9): 105-114. |
| [2] | 张超超, 韩开元, 王彤, 陈仲. 毛白杨PtoYABBY2和PtoYABBY12的克隆及功能分析[J]. 生物技术通报, 2025, 41(9): 256-264. |
| [3] | 史发超, 姜永华, 刘海伦, 文英杰, 严倩. 荔枝LcTFL1基因的克隆与功能分析[J]. 生物技术通报, 2025, 41(9): 159-167. |
| [4] | 董向向, 缪百灵, 许贺娟, 陈娟娟, 李亮杰, 龚守富, 朱庆松. 森林草莓FveBBX20基因的生物信息学分析及开花调控功能[J]. 生物技术通报, 2025, 41(9): 115-123. |
| [5] | 王斌, 林冲, 袁晓, 蒋园园, 王玉昆, 肖艳辉. bHLH转录因子UNE10克隆及其在丁香罗勒挥发性化合物合成调控中的功能[J]. 生物技术通报, 2025, 41(9): 207-218. |
| [6] | 黄诗宇, 田姗姗, 杨天为, 高曼熔, 张尚文. 赤苍藤WRI1基因家族的全基因组鉴定及表达模式分析[J]. 生物技术通报, 2025, 41(8): 242-254. |
| [7] | 李开杰, 吴瑶, 李丹丹. 红花CtbHLH128基因克隆及调控干旱胁迫应答功能研究[J]. 生物技术通报, 2025, 41(8): 234-241. |
| [8] | 曾丹, 黄园, 王健, 张艳, 刘庆霞, 谷荣辉, 孙庆文, 陈宏宇. 铁皮石斛bZIP转录因子家族全基因组鉴定及表达分析[J]. 生物技术通报, 2025, 41(8): 197-210. |
| [9] | 牛景萍, 赵婧, 郭茜, 王书宏, 赵晋忠, 杜维俊, 殷丛丛, 岳爱琴. 基于WGCNA鉴定大豆抗大豆花叶病毒NAC转录因子及其诱导表达分析[J]. 生物技术通报, 2025, 41(7): 95-105. |
| [10] | 李霞, 张泽伟, 刘泽军, 王楠, 郭江波, 辛翠花, 张彤, 简磊. 马铃薯转录因子StMYB96的克隆及功能研究[J]. 生物技术通报, 2025, 41(7): 181-192. |
| [11] | 魏雨佳, 李岩, 康语涵, 弓晓楠, 杜敏, 涂岚, 石鹏, 于子涵, 孙彦, 张昆. 白颖苔草CrMYB4基因的克隆和表达分析[J]. 生物技术通报, 2025, 41(7): 248-260. |
| [12] | 蒋天威, 马培杰, 李亚娇, 陈才俊, 刘晓霞, 王小利. 二穗短柄草对光周期的代谢响应分析[J]. 生物技术通报, 2025, 41(7): 237-247. |
| [13] | 李锐, 胡婷, 陈树溦, 王尧, 王计平. 紫苏PfMYB80转录因子正向调控花青素的生物合成[J]. 生物技术通报, 2025, 41(6): 243-255. |
| [14] | 郭涛, 艾丽皎, 邹世慧, 周玲, 李学梅. 山茶CjRAV1调控开花延迟的功能研究[J]. 生物技术通报, 2025, 41(6): 208-217. |
| [15] | 程珊, 王会伟, 陈晨, 朱雅婧, 李春鑫, 别海, 王树峰, 陈献功, 张向歌. 油莎豆MYB转录因子基因CeMYB154克隆及耐盐功能分析[J]. 生物技术通报, 2025, 41(6): 218-228. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||