







生物技术通报 ›› 2026, Vol. 42 ›› Issue (1): 294-304.doi: 10.13560/j.cnki.biotech.bull.1985.2024-1253
刘长命1( ), 张新悦1, 杨昕萌1, 刘阳1, 李玥涵1, 汪可清2
), 张新悦1, 杨昕萌1, 刘阳1, 李玥涵1, 汪可清2
收稿日期:2024-12-24
出版日期:2026-01-26
发布日期:2026-02-04
作者简介:刘长命,男,博士,副教授,研究方向 :园艺植物种质资源应用;E-mail: liujie2061@163.com;刘长命同为本文
基金资助:
LIU Chang-ming1(), ZHANG Xin-yue1, YANG Xin-meng1, LIU Yang1, LI Yue-han1, WANG Ke-qing2
Received:2024-12-24
Published:2026-01-26
Online:2026-02-04
摘要:
目的 从魔芋球茎中首次分离到1株软腐病致病菌株(Raoultella ornithinolytica Q-8,登录号:CP173279),从全基因组层面对其进行序列分析和基因功能注释,为深入研究该菌株的致病机理和对其有效防治提供理论依据。 方法 利用3代全基因组测序技术,通过Canu、Prodigal、RepeatMasker等软件对基因组数据进行组装和基因预测,利用KEGG、PHI-base等12个数据库进行功能注释和对比分析,并根据基因组ORF序列利用FastTree2构建系统发育树。 结果 R. ornithinolytica Q-8基因组总长度为5.44 Mb,GC含量55.9%,含有4 962个编码基因、6个假基因、11个基因岛、25个rRNA、129个其他ncRNA及16种不同类型的CRISPR系统。其中有4 833个基因被注释到22个基因簇中,3 126个基因被富集到114条代谢通路中,有1 714个运输蛋白相关基因、18个抗生素耐药相关基因、1 854个病原体宿主互作相关基因和1 067个毒力因子相关基因。系统发育树分析结果表明,R. ornithinolytica Q-8与其他8株R. ornithinolytica聚在一大分支,而与已报道的2种魔芋软腐病病原菌P. carotovorum和D. chrysanthemi距离较远。 结论 R. ornithinolytica Q-8是1株新的魔芋软腐病致病菌,其基因组中存在多个抗生素耐药基因和病原菌宿主互作基因,这些基因可能在其致病中发挥着重要作用。
刘长命, 张新悦, 杨昕萌, 刘阳, 李玥涵, 汪可清. 一株新的魔芋软腐病致病菌的生物学特性及基因组分析[J]. 生物技术通报, 2026, 42(1): 294-304.
LIU Chang-ming, ZHANG Xin-yue, YANG Xin-meng, LIU Yang, LI Yue-han, WANG Ke-qing. Biological Characteristics and Genomic Analysis of a Novel Pathogen Strain Raoultella ornithinolytica Causing Soft Rot Disease in Konjac[J]. Biotechnology Bulletin, 2026, 42(1): 294-304.
| 样本编号 Sample No. | 采样地点 Sampling location | 经度 Longitude (E) | 纬度 Latitude (N) | 海拔 Elevation (m) |
|---|---|---|---|---|
| 1 | 丹凤县南丈沟村 Nanzhanggou village, Danfeng county | 110°60′23″ | 33°16′81″ | 572 |
| 2 | 丹凤县南丈沟村 Nanzhanggou village, Danfeng county | 110°59′70″ | 33°16′71″ | 560 |
| 3 | 山阳县阳河村 Yanghe village, Shangyang county | 110°22′38″ | 34°47′56″ | 770 |
| 4 | 山阳县阳河村 Yanghe village, Shangyang county | 110°22′62″ | 34°47′65″ | 756 |
| 5 | 商南县小河村 Xiaohe village, Shangnan county | 110°46′22″ | 33°47′46″ | 438 |
| 6 | 商南县小河村 Xiaohe village, Shangnan county | 110°46′25″ | 33°47′44″ | 430 |
表1 魔芋软腐病病株样本采样地信息
Table 1 The sites information of konjac (Amorphophallus spp. ) plant samples with soft rot symptoms
| 样本编号 Sample No. | 采样地点 Sampling location | 经度 Longitude (E) | 纬度 Latitude (N) | 海拔 Elevation (m) |
|---|---|---|---|---|
| 1 | 丹凤县南丈沟村 Nanzhanggou village, Danfeng county | 110°60′23″ | 33°16′81″ | 572 |
| 2 | 丹凤县南丈沟村 Nanzhanggou village, Danfeng county | 110°59′70″ | 33°16′71″ | 560 |
| 3 | 山阳县阳河村 Yanghe village, Shangyang county | 110°22′38″ | 34°47′56″ | 770 |
| 4 | 山阳县阳河村 Yanghe village, Shangyang county | 110°22′62″ | 34°47′65″ | 756 |
| 5 | 商南县小河村 Xiaohe village, Shangnan county | 110°46′22″ | 33°47′46″ | 438 |
| 6 | 商南县小河村 Xiaohe village, Shangnan county | 110°46′25″ | 33°47′44″ | 430 |
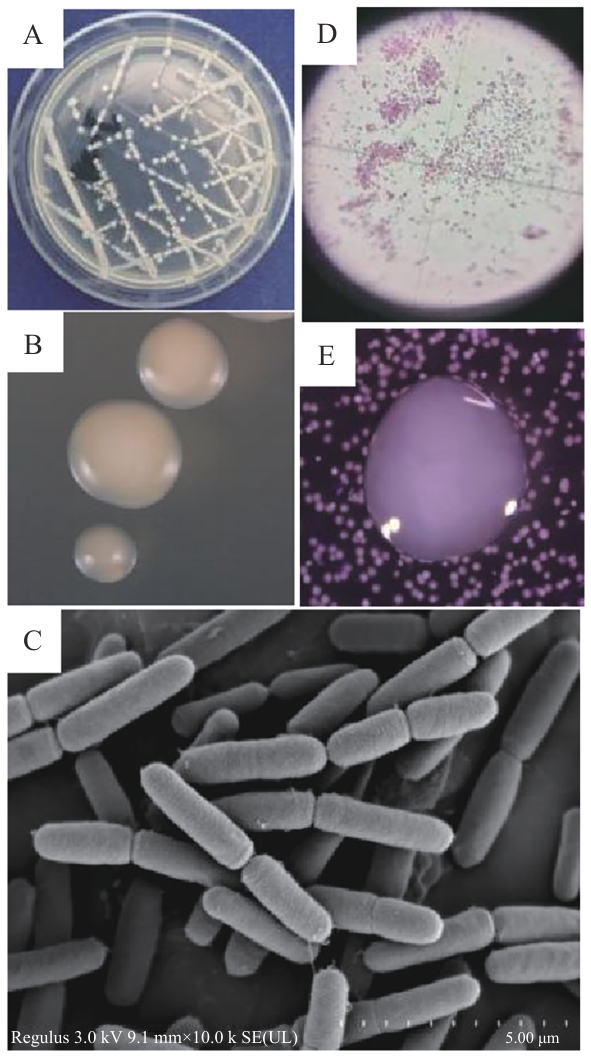
图1 病原菌R. ornithinolytica Q-8的菌落和细胞形态特征A:LB平板上的菌落形态;B:体式显微镜下的菌落形态;C:扫描电镜下的细胞形态;D:革兰氏染色结果;E:CVP平板上的菌落形态
Fig. 1 Colonies and cell morphologies of pathogen R. ornithinolytica Q-8A: Colony on LB plate. B: Colony under stereo microscope. C: Cells under scanning electron microscope. D: Gram staining. E: Colony on CVP plate
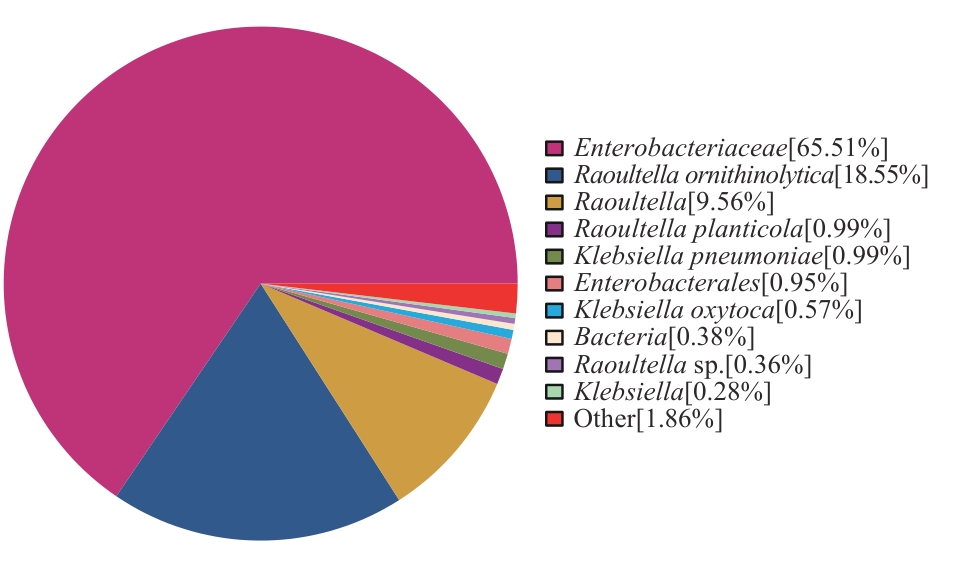
图2 R. ornithinolytica Q-8基因组NR数据库比对的物种分布图
Fig. 2 Species distribution map of strain R. ornithinolytica Q-8 genome from the NR database alignment
菌株 Strain | 基因组大小 Genome length (bp) | 编码基因 Coding genes | GC 含量 GC content (%) | 假基因 Pseudo genes | rRNA | tRNA | ncRNAs | 收集日期 Collecting date | 来源 Source | 来源地 Source location |
|---|---|---|---|---|---|---|---|---|---|---|
| Q-8 | 5 440 241 | 4 962 | 55.9 | 6 | 25 | 87 | 129 | 2022.04 | 魔芋 Konjac | 中国商洛 Shangluo, China |
| NY1 | 5 592 869 | 5 195 | 55.5 | 118 | 25 | 85 | 12 | 2019.08 | 尿液 Urine | 中国济南 Jinan, China |
| GSH0205-8M-1 | 5 490 896 | 5 540 | 54.7 | 118 | 25 | 82 | 11 | 2019.02 | 污水处理厂出水 Wastewater treatment plant effluent | 日本东京 Tokyo, Japan |
| FDAARGOS 431 | 5 633 990 | 5 600 | 55.5 | 225 | 25 | 85 | 12 | 2015.07 | 病人直肠 The patient’s rectum | 加拿大不列颠哥伦比亚 British Columbia, Canada |
| SECR20-0777 | 5 460 546 | 5 088 | 56.0 | 64 | 25 | 85 | 13 | 2020.03 | 碳青霉烯类耐药肠杆菌科细菌感染人直肠 CRE infection on human rectal | 韩国首尔 Seoul, Korea |
表2 五株解鸟氨酸拉乌尔菌基因组的一般特征
Table 2 General characteristics of the five R. ornithinolytica genomes
菌株 Strain | 基因组大小 Genome length (bp) | 编码基因 Coding genes | GC 含量 GC content (%) | 假基因 Pseudo genes | rRNA | tRNA | ncRNAs | 收集日期 Collecting date | 来源 Source | 来源地 Source location |
|---|---|---|---|---|---|---|---|---|---|---|
| Q-8 | 5 440 241 | 4 962 | 55.9 | 6 | 25 | 87 | 129 | 2022.04 | 魔芋 Konjac | 中国商洛 Shangluo, China |
| NY1 | 5 592 869 | 5 195 | 55.5 | 118 | 25 | 85 | 12 | 2019.08 | 尿液 Urine | 中国济南 Jinan, China |
| GSH0205-8M-1 | 5 490 896 | 5 540 | 54.7 | 118 | 25 | 82 | 11 | 2019.02 | 污水处理厂出水 Wastewater treatment plant effluent | 日本东京 Tokyo, Japan |
| FDAARGOS 431 | 5 633 990 | 5 600 | 55.5 | 225 | 25 | 85 | 12 | 2015.07 | 病人直肠 The patient’s rectum | 加拿大不列颠哥伦比亚 British Columbia, Canada |
| SECR20-0777 | 5 460 546 | 5 088 | 56.0 | 64 | 25 | 85 | 13 | 2020.03 | 碳青霉烯类耐药肠杆菌科细菌感染人直肠 CRE infection on human rectal | 韩国首尔 Seoul, Korea |
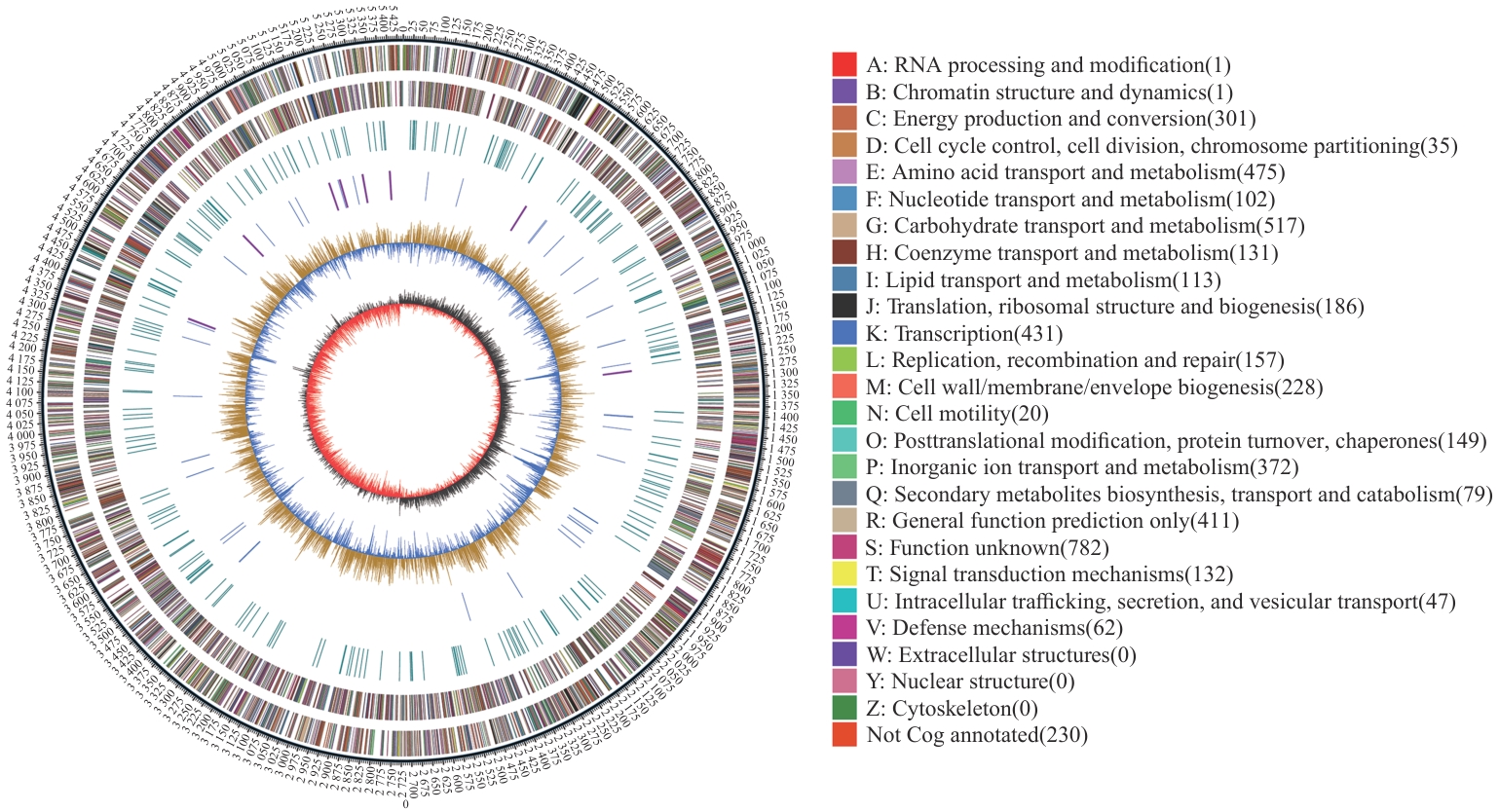
图3 R. ornithinolytica Q-8的全基因组图形图谱
Fig. 3 Circular mapping of the whole genome of R. ornithinolytica Q-8
CRISPR编号 CRISPR ID | 起始位置 Start | 结束位置 End | 重复次数 Repeat number | 平均重复长度 Average repeat length (bp) | 间隔序列数量 Spacer number | 平均间隔序列长度 Average spacer length (bp) |
|---|---|---|---|---|---|---|
| CRISPR.1 | 347 753 | 348 100 | 8 | 26 | 7 | 20 |
| CRISPR.2 | 1 100 503 | 1 100 601 | 2 | 33 | 1 | 33 |
| CRISPR.3 | 1 196 948 | 1 197 054 | 3 | 19 | 2 | 25 |
| CRISPR.4 | 2 464 743 | 2 464 894 | 3 | 38 | 2 | 19 |
| CRISPR.5 | 2 595 049 | 2 595 128 | 2 | 26 | 1 | 28 |
| CRISPR.6 | 2 595 022 | 2 595 269 | 5 | 26 | 4 | 29 |
| CRISPR.7 | 2 901 354 | 2 901 775 | 9 | 30 | 8 | 19 |
| CRISPR.8 | 3 255 068 | 3 255 128 | 2 | 21 | 1 | 19 |
| CRISPR.9 | 3 316 575 | 3 316 825 | 4 | 35 | 3 | 37 |
| CRISPR.10 | 3 608 294 | 3 608 756 | 7 | 31 | 6 | 41 |
| CRISPR.11 | 3 752 358 | 3 752 470 | 2 | 32 | 1 | 49 |
| CRISPR.12 | 3 969 172 | 3 969 472 | 4 | 37 | 3 | 51 |
| CRISPR.13 | 4 705 327 | 4 705 595 | 5 | 38 | 4 | 19 |
| CRISPR.14 | 4 846 212 | 4 846 348 | 3 | 33 | 2 | 19 |
| CRISPR.15 | 4 853 717 | 4 853 915 | 3 | 31 | 2 | 53 |
| CRISPR.16 | 5 039 222 | 5 039 506 | 5 | 21 | 4 | 45 |
表3 R. ornithinolytica Q-8的CRISPR预测结果统计
Table 3 Statistics of CRISPR predicting results of R. ornithinolytica Q-8
CRISPR编号 CRISPR ID | 起始位置 Start | 结束位置 End | 重复次数 Repeat number | 平均重复长度 Average repeat length (bp) | 间隔序列数量 Spacer number | 平均间隔序列长度 Average spacer length (bp) |
|---|---|---|---|---|---|---|
| CRISPR.1 | 347 753 | 348 100 | 8 | 26 | 7 | 20 |
| CRISPR.2 | 1 100 503 | 1 100 601 | 2 | 33 | 1 | 33 |
| CRISPR.3 | 1 196 948 | 1 197 054 | 3 | 19 | 2 | 25 |
| CRISPR.4 | 2 464 743 | 2 464 894 | 3 | 38 | 2 | 19 |
| CRISPR.5 | 2 595 049 | 2 595 128 | 2 | 26 | 1 | 28 |
| CRISPR.6 | 2 595 022 | 2 595 269 | 5 | 26 | 4 | 29 |
| CRISPR.7 | 2 901 354 | 2 901 775 | 9 | 30 | 8 | 19 |
| CRISPR.8 | 3 255 068 | 3 255 128 | 2 | 21 | 1 | 19 |
| CRISPR.9 | 3 316 575 | 3 316 825 | 4 | 35 | 3 | 37 |
| CRISPR.10 | 3 608 294 | 3 608 756 | 7 | 31 | 6 | 41 |
| CRISPR.11 | 3 752 358 | 3 752 470 | 2 | 32 | 1 | 49 |
| CRISPR.12 | 3 969 172 | 3 969 472 | 4 | 37 | 3 | 51 |
| CRISPR.13 | 4 705 327 | 4 705 595 | 5 | 38 | 4 | 19 |
| CRISPR.14 | 4 846 212 | 4 846 348 | 3 | 33 | 2 | 19 |
| CRISPR.15 | 4 853 717 | 4 853 915 | 3 | 31 | 2 | 53 |
| CRISPR.16 | 5 039 222 | 5 039 506 | 5 | 21 | 4 | 45 |
| 通用数据库 General database | 基因数量 Number of genes | 专用数据库 Special database | 基因数量 Number of genes |
|---|---|---|---|
| eggNOG | 4 732 | CAZy | 166 |
| GO | 3 124 | TCDB | 1 714 |
| KEGG | 3 342 | PHI-base | 1 854 |
| NR | 4 949 | CARD | 18 |
| Pfam | 4 648 | VFDB | 1 067 |
| SwissProt | 4 019 | ||
| TrEMBL | 4 946 |
表4 R. ornithinolytica Q-8基因组功能分析汇总
Table 4 Summary of genome function analysis of R. ornithinolytica Q-8
| 通用数据库 General database | 基因数量 Number of genes | 专用数据库 Special database | 基因数量 Number of genes |
|---|---|---|---|
| eggNOG | 4 732 | CAZy | 166 |
| GO | 3 124 | TCDB | 1 714 |
| KEGG | 3 342 | PHI-base | 1 854 |
| NR | 4 949 | CARD | 18 |
| Pfam | 4 648 | VFDB | 1 067 |
| SwissProt | 4 019 | ||
| TrEMBL | 4 946 |
| 抗生素抗性 Antibiotic resistance | 基因列表 Gene list |
|---|---|
| 氨基香豆素类 Aminocoumarin | GE001580; GE001581 |
| 氨基糖苷类 Aminoglycoside | GE001289 |
| 碳青霉烯类、头孢菌素类、单环β-内酰胺类、青霉素类、头霉素类 Carbapenem, cephalosporin, monobactam, penam, cephamycin | GE004174 |
| 埃尔法霉素类 Elfamycin | GE000413; GE004752 |
| 氟喹诺酮类 Fluoroquinolone | GE001066; GE001068 |
| 磷霉素 Fosfomycin | GE000039; GE004353 |
| 大环内酯类 Macrolide | GE002389 |
| 硝基咪唑类 Nitroimidazole | GE003296 |
| 青霉素类、大环内酯类、氟喹诺酮类 Penam, macrolide, fluoroquinolone | GE000393 |
| 青霉素类、三氯生、苯酚类、头孢菌素类、四环素类、甘氨环素类、利福霉素类、氟喹诺酮类 Penam, triclosan, phenicol, cephalosporin, tetracycline, glycylcycline, rifamycin, fluoroquinolone | GE002486 |
| 青霉素类、三氯生、四环素类、头孢菌素类、甘氨环素类、氟喹诺酮类、利福霉素类、苯酚类 Penam, triclosan, tetracycline, cephalosporin, glycylcycline, fluoroquinolone, rifamycin, phenicol | GE003804 |
| 青霉烯类、四环素类、头孢菌素类、碳青霉烯类、青霉素类、三氯生、头霉素类、甘氨环素类、单环β-内酰胺类、氟喹诺酮类、苯酚类、利福霉素类 Penem, tetracycline, cephalosporin, carbapenem, penam, triclosan, cephamycin, glycylcycline, monobactam, fluoroquinolone, phenicol, rifamycin | GE002485 |
| 肽类 Peptide | GE000273 |
| 四环素类、氟喹诺酮类 Tetracycline, fluoroquinolone | GE001127 |
表5 R. ornithinolytica Q-8基因组中的抗生素抗性蛋白种类
Table 5 Types of antibiotic resistance proteins in the genome of R. ornithinolytica Q-8
| 抗生素抗性 Antibiotic resistance | 基因列表 Gene list |
|---|---|
| 氨基香豆素类 Aminocoumarin | GE001580; GE001581 |
| 氨基糖苷类 Aminoglycoside | GE001289 |
| 碳青霉烯类、头孢菌素类、单环β-内酰胺类、青霉素类、头霉素类 Carbapenem, cephalosporin, monobactam, penam, cephamycin | GE004174 |
| 埃尔法霉素类 Elfamycin | GE000413; GE004752 |
| 氟喹诺酮类 Fluoroquinolone | GE001066; GE001068 |
| 磷霉素 Fosfomycin | GE000039; GE004353 |
| 大环内酯类 Macrolide | GE002389 |
| 硝基咪唑类 Nitroimidazole | GE003296 |
| 青霉素类、大环内酯类、氟喹诺酮类 Penam, macrolide, fluoroquinolone | GE000393 |
| 青霉素类、三氯生、苯酚类、头孢菌素类、四环素类、甘氨环素类、利福霉素类、氟喹诺酮类 Penam, triclosan, phenicol, cephalosporin, tetracycline, glycylcycline, rifamycin, fluoroquinolone | GE002486 |
| 青霉素类、三氯生、四环素类、头孢菌素类、甘氨环素类、氟喹诺酮类、利福霉素类、苯酚类 Penam, triclosan, tetracycline, cephalosporin, glycylcycline, fluoroquinolone, rifamycin, phenicol | GE003804 |
| 青霉烯类、四环素类、头孢菌素类、碳青霉烯类、青霉素类、三氯生、头霉素类、甘氨环素类、单环β-内酰胺类、氟喹诺酮类、苯酚类、利福霉素类 Penem, tetracycline, cephalosporin, carbapenem, penam, triclosan, cephamycin, glycylcycline, monobactam, fluoroquinolone, phenicol, rifamycin | GE002485 |
| 肽类 Peptide | GE000273 |
| 四环素类、氟喹诺酮类 Tetracycline, fluoroquinolone | GE001127 |
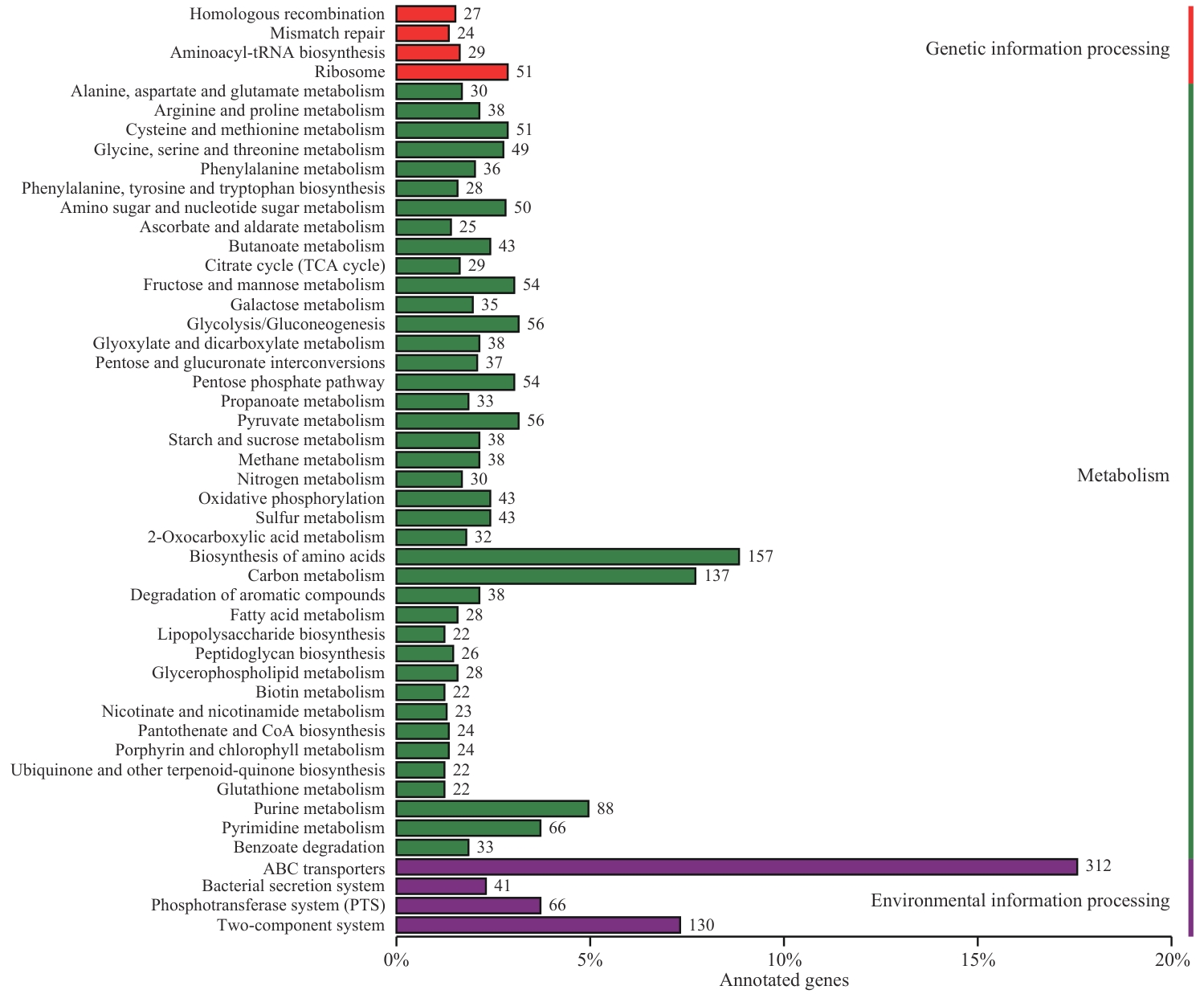
图4 R. ornithinolytica Q-8基因组KEGG数据库主要代谢通路分析
Fig. 4 Major metabolic pathway analysis of R. ornithinolytica Q-8 genome in KEGG database
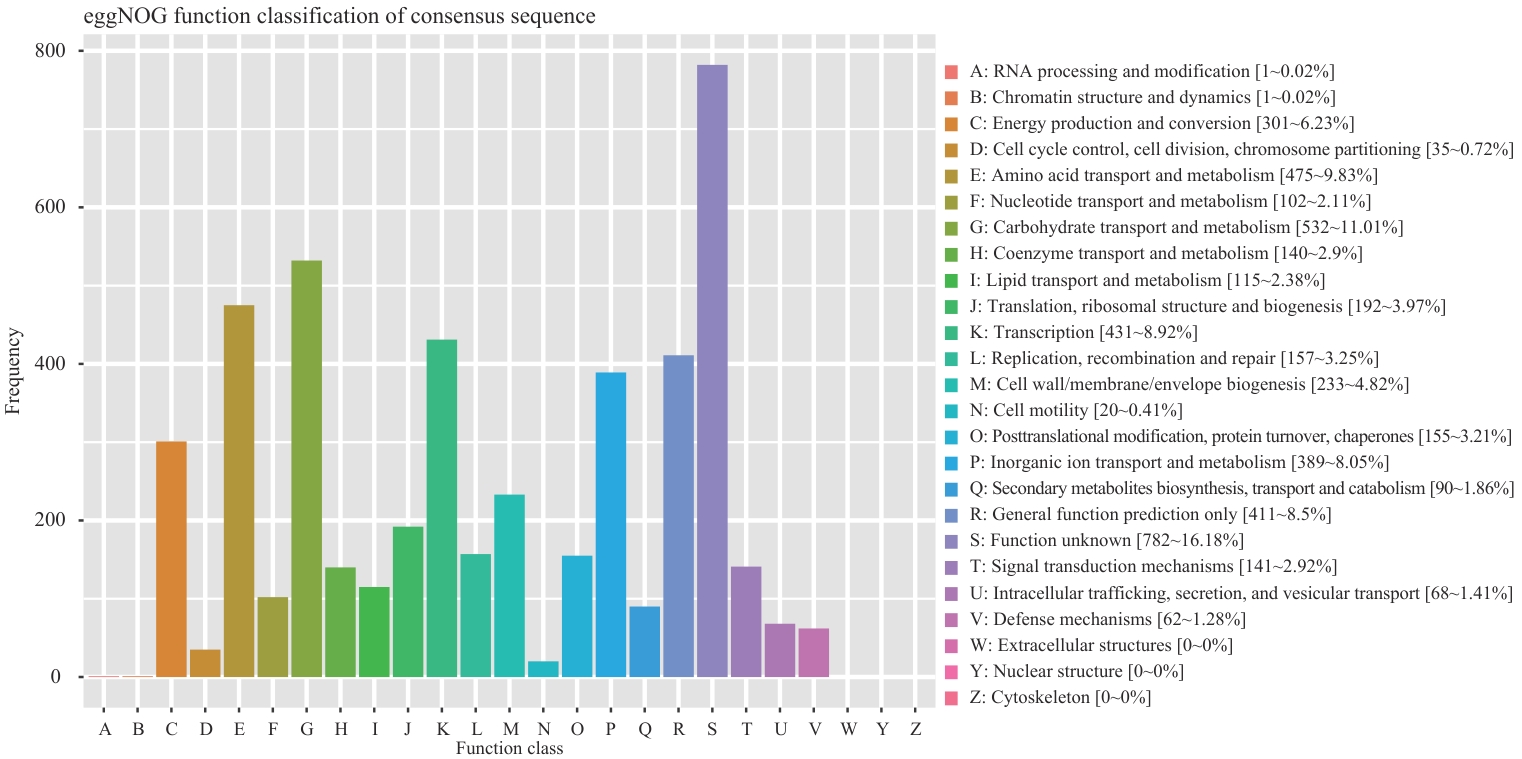
图5 R. ornithinolytica Q-8基因组eggNOG数据库同源基因簇分析
Fig. 5 Homologous gene clusters analysis of R. ornithinolytica Q-8 genome in eggNOG database
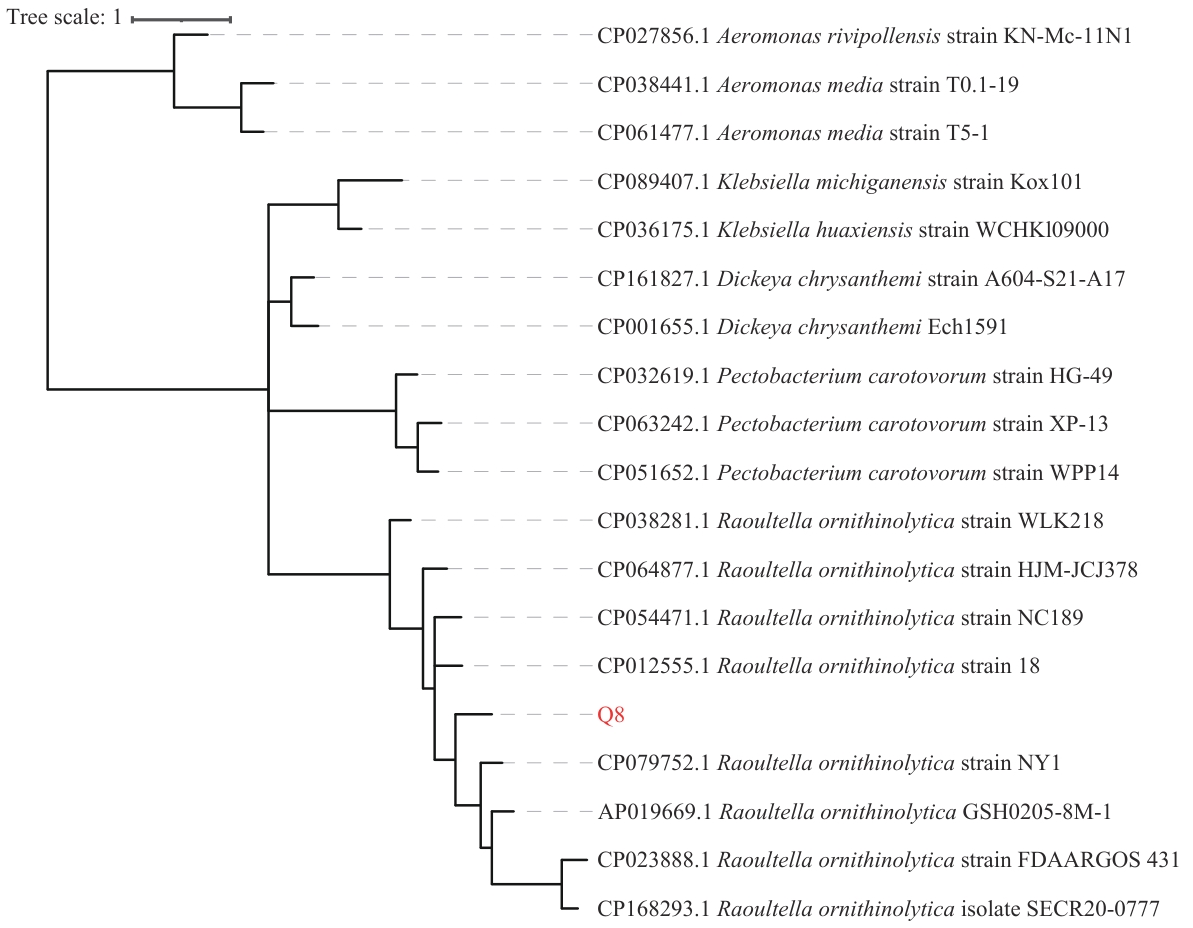
图6 R. ornithinolytica Q-8菌株基于ORF序列的进化树分析
Fig. 6 Phylogenetic tree analysis of R. ornithinolytica Q-8strains based on ORF sequences
| [1] | 吴金平. 魔芋软腐病病原菌及其拮抗菌的研究 [D]. 武汉: 武汉大学, 2010. |
| Wu JP. Pathogens and antagonistic bacteria of soft rot disease on konjac [D]. Wuhan: Wuhan University, 2010. | |
| [2] | 孙苗苗. 魔芋软腐病病原鉴定及快速检测技术研究 [D]. 武汉: 华中农业大学, 2019. |
| Sun MM. Pathogen identification and rapid detection method development for soft rot of Amorphophallus konjac [D]. Wuhan: Huazhong Agricultural University, 2019. | |
| [3] | 池丹, 胡燕燕, 马继华, 等. 一株解鸟氨酸拉乌尔菌的鉴定及其β内酰胺酶编码基因的研究 [J]. 中华检验医学杂志, 2013, 36(11): 1045-1047. |
| Chi D, Hu YY, Ma JH, et al. Identification of a Raoul strain of ornithine and study on its β-lactamase coding gene [J]. Chin J Lab Med, 2013, 36(11): 1045-1047. | |
| [4] | Campos J, Mourão J, Pestana N, et al. Microbiological quality of ready-to-eat salads: an underestimated vehicle of bacteria and clinically relevant antibiotic resistance genes [J]. Int J Food Microbiol, 2013, 166(3): 464-470. |
| [5] | Lam PW, Salit IE. Raoultella planticola bacteremia following consumption of seafood [J]. Can J Infect Dis Med Microbiol, 2014, 25(4): e83-e84. |
| [6] | 黄敏, 何鹏搏, 何鹏飞, 等. 肺炎克雷伯氏菌(Klebsiella pneumoniae)对植物影响的研究进展 [J]. 植物病理学报, 2022, 52(4): 511-521. |
| Huang M, He PB, He PF, et al. Research advances in effects of Klebsiella pneumoniae on plants [J]. Acta Phytopathol Sin, 2022, 52(4): 511-521. | |
| [7] | Seng P, Boushab BM, Romain F, et al. Emerging role of Raoultella ornithinolytica in human infections: a series of cases and review of the literature [J]. Int J Infect Dis, 2016, 45: 65-71. |
| [8] | 海丹, 黄现青, 乔明武, 等. 临期盒装豆腐腐败菌鉴定及抑菌效果评价 [J]. 河南农业大学学报, 2024, 58(2): 298-306. |
| Hai D, Huang XQ, Qiao MW, et al. Identification of spoilage bacteria in deadline boxed tofu and evaluation of their inhibitory effects [J]. J Henan Agric Univ, 2024, 58(2): 298-306. | |
| [9] | 原小迪, 李钰, 陈艳芳, 等. 肺炎克雷伯氏菌环境菌株血清型及对玉米的致病性测定 [J]. 云南农业大学学报: 自然科学, 2022, 37(3): 422-428. |
| Yuan XD, Li Y, Chen YF, et al. Serotype of Klebsiella pneumoniae environmental strain and its pathogenicity to maize [J]. J Yunnan Agric Univ Nat Sci, 2022, 37(3): 422-428. | |
| [10] | 魏超, 代晓航, 郭灵安, 等. 利用MALDI-TOF MS鉴定芽苗菜中解鸟氨酸拉乌尔菌和肺炎克雷伯菌的研究 [J]. 分析测试学报, 2018, 37(1): 76-81. |
| Wei C, Dai XH, Guo LA, et al. Research on identification of Raoultella ornithinolytica and Klebsiella pneumoniae in sprouts by MALDI-TOF MS [J]. J Instrum Anal, 2018, 37(1): 76-81. | |
| [11] | 邱睿, 李成军, 陈玉国, 等. 引起烟苗软腐的病原鉴定及致病性分析 [J]. 烟草科技, 2018, 51(8): 22-26. |
| Qiu R, Li CJ, Chen YG, et al. Identification and pathogenicity analysis of soft rot in tobacco seedlings [J]. Tob Sci Technol, 2018, 51(8): 22-26. | |
| [12] | 方中达. 植病研究方法 [M]. 3版. 北京: 中国农业出版社, 1998. |
| Fang ZD. Research methods of plant diseases [M]. 3rd ed. Beijing: China Agriculture Press, 1998. | |
| [13] | 肖媛, 刘伟, 汪艳, 等. 生物样品的扫描电镜制样干燥方法 [J]. 实验室研究与探索, 2013, 32(5): 45-53, 172. |
| Xiao Y, Liu W, Wang Y, et al. Drying methods of biological sample preparation for scanning electron microscope [J]. Res Explor Lab, 2013, 32(5): 45-53, 172. | |
| [14] | McClinton B, Crinnion LA, McKibbin M, et al. Targeted nanopore sequencing enables complete characterisation of structural deletions initially identified using exon-based short-read sequencing strategies [J]. Mol Genet Genom Med, 2023, 11(6): e2164. |
| [15] | Koren S, Walenz BP, Berlin K, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation [J]. Genome Res, 2017, 27(5): 722-736. |
| [16] | Vaser R, Sović I, Nagarajan N, et al. Fast and accurate de novo genome assembly from long uncorrected reads [J]. Genome Res, 2017, 27(5): 737-746. |
| [17] | Hunt M, De Silva N, Otto TD, et al. Circlator: automated circularization of genome assemblies using long sequencing reads [J]. Genome Biol, 2015, 16: 294. |
| [18] | Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement [J]. PLoS One, 2014, 9(11): e112963. |
| [19] | Hyatt D, Chen GL, Locascio PF, et al. Prodigal: prokaryotic gene recognition and translation initiation site identification [J]. BMC Bioinformatics, 2010, 11: 119. |
| [20] | Chen NS. Using RepeatMasker to identify repetitive elements in genomic sequences [J]. Curr Protoc Bioinform, 2004, 5(1): 4.10.1-4.10.14. |
| [21] | Chan PP, Lowe TM. tRNAscan-SE searching for tRNA genes in genomic sequences [J]. Methods Mol Biol, 2019, 1962: 1-14. |
| [22] | Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches [J]. Bioinformatics, 2013, 29(22): 2933-2935. |
| [23] | Bland C, Ramsey TL, Sabree F, et al. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats [J]. BMC Bioinformatics, 2007, 8: 209. |
| [24] | Bertelli C, Brinkman FSL. Improved genomic island predictions with IslandPath-DIMOB [J]. Bioinformatics, 2018, 34(13): 2161-2167. |
| [25] | Blin K, Shaw S, Steinke K, et al. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline [J]. Nucleic Acids Res, 2019, 47(W1): W81-W87. |
| [26] | Ashburner M, Ball CA, Blake JA, et al. Gene Ontology: tool for the unification of biology [J]. Nat Genet, 2000, 25(1): 25-29. |
| [27] | Finn RD, Bateman A, Clements J, et al. Pfam: the protein families database [J]. Nucleic Acids Res, 2014, 42(Database issue): D222-D230. |
| [28] | Bairoch A, Apweiler R. The SWISS-PROT protein sequence data bank and its new supplement TREMBL [J]. Nucleic Acids Res, 1996, 24(1): 21-25. |
| [29] | Cantarel BL, Coutinho PM, Rancurel C, et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics [J]. Nucleic Acids Res, 2009, 37(Database issue): D233-D238. |
| [30] | Jia BF, Raphenya AR, Alcock B, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database [J]. Nucleic Acids Res, 2017, 45(D1): D566-D573. |
| [31] | Winnenburg R, Baldwin TK, Urban M, et al. PHI-base: a new database for pathogen host interactions [J]. Nucleic Acids Res, 2006, 34(Database issue): D459-D464. |
| [32] | Chen LH, Yang J, Yu J, et al. VFDB: a reference database for bacterial virulence factors [J]. Nucleic Acids Res, 2005, 33(Database issue): D325-D328. |
| [33] | Powell S, Forslund K, Szklarczyk D, et al. eggNOG v4.0: nested orthology inference across 3686 organisms [J]. Nucleic Acids Res, 2014, 42(Database issue): D231-D239. |
| [34] | Saier MH Jr, Tran CV, Barabote RD. TCDB: the Transporter Classification Database for membrane transport protein analyses and information [J]. Nucleic Acids Res, 2006, 34(Database issue): D181-D186. |
| [35] | Sun JH, Lu F, Luo YJ, et al. OrthoVenn3: an integrated platform for exploring and visualizing orthologous data across genomes [J]. Nucleic Acids Res, 2023, 51(W1): W397-W403. |
| [36] | Ebmeyer S, Coertze RD, Berglund F, et al. GEnView: a gene-centric, phylogeny-based comparative genomics pipeline for bacterial genomes and plasmids [J]. Bioinformatics, 2022, 38(6): 1727-1728. |
| [37] | Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND [J]. Nat Methods, 2015, 12(1): 59-60. |
| [38] | Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix [J]. Mol Biol Evol, 2009, 26(7): 1641-1650. |
| [39] | Katoh K, Misawa K, Kuma KI, et al. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform [J]. Nucleic Acids Res, 2002, 30(14): 3059-3066. |
| [40] | Li WZ, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences [J]. Bioinformatics, 2006, 22(13): 1658-1659. |
| [41] | Ma B, Hibbing ME, Kim HS, et al. Host range and molecular phylogenies of the soft rot enterobacterial genera Pectobacterium and Dickeya [J]. Phytopathology, 2007, 97(9): 1150-1163. |
| [42] | Nabhan S, De Boer SH, Maiss E, et al. Pectobacterium aroidearum sp. nov., a soft rot pathogen with preference for monocotyledonous plants [J]. Int J Syst Evol Microbiol, 2013, 63(Pt 7): 2520-2525. |
| [43] | 黄俊斌, 邱仁胜, 赵纯森, 等. 魔芋软腐病病原菌的鉴定及生物学特性初步研究[J]. 华中农业大学学报, 1999, 18(5): 413-415. |
| Huang JB, Qiu RS, Zhao CS, et al. Identification and preliminary study on the biological characteristics of the pathogen of konjac soft rot disease[J]. J Huazhong Agric Univ, 1999, 18(5): 413-415. | |
| [44] | 徐炜. 岚皋县魔芋软腐病和白绢病病原菌的分离鉴定和生物防控初探 [D]. 杨凌: 西北农林科技大学, 2011. |
| Xu W. Isolation and identification of the soft rot and Sclerotium rolfsii of konjac and study of biological control in Langao county [D]. Yangling: Northwest A & F University, 2011. | |
| [45] | 唐嘉义, 张泽. 魔芋软腐病病原菌鉴定及部分生物学特性研究 [J]. 云南农业大学学报, 2001, 16(3): 185-187. |
| Tang JY, Zhang Z. Studies on partial biological character and identification of bacterial soft rot of elephant taro [J]. J Yunnan Agric Univ, 2001, 16(3): 185-187. | |
| [46] | 沈业寿, 储苏. 魔芋软腐病病原菌的分离及致病性研究 [J]. 安徽大学学报: 自然科学版, 2002, 26(1): 96-100. |
| Shen YS, Chu S. Studies on the pathogenicity and the isolation of pathogen from konnyaku’s soft rot [J]. J Anhui Univ Nat Sci Ed, 2002, 26(1): 96-100. | |
| [47] | 修建华, 姬广海, 王敏, 等. 魔芋软腐病菌分子鉴定与遗传多样性 [J]. 微生物学报, 2006, 46(4): 522-525. |
| Xiu JH, Ji GH, Wang M, et al. Molecular identification and genetic diversity in Konnyaku’s soft rot bacteria [J]. Acta Microbiol Sin, 2006, 46(4): 522-525. | |
| [48] | 刘长命, 覃晓冉, 汪可清, 等. 解鸟氨酸拉乌尔菌引起魔芋软腐病的首次报道 [J]. 植物病理学报, 2025, 55(3): 545-549. |
| Liu CM, Qin XR, Wang KQ, et al. First report of Raoultella ornithinolytica causing soft rot on konjac [J]. Acta Phytopathol Sin, 2025, 55(3): 545-549. | |
| [49] | 唐昊, 孙灿, 李沅秋, 等. 纤维素降解菌Raoultella ornithinolytica LL1的筛选及基因组测序 [J]. 生物技术通报, 2021, 37(6): 85-96. |
| Tang H, Sun C, Li YQ, et al. Screening and genome sequencing of cellulytic bacterium Raoultella ornithinolytica LL1 [J]. Biotechnol Bull, 2021, 37(6): 85-96. | |
| [50] | Soanker R, Vemu L, Vimala S, et al. The effect of double carbapenem regimen in the management of carbapenem-resistant Klebsiella pneumoniae infections: a report of five cases [J]. Cureus, 2024, 16(1): e52023. |
| [51] | 冯洁. 植物病原细菌分类最新进展 [J]. 中国农业科学, 2017, 50(12): 2305-2314. |
| Feng J. Recent advances in taxonomy of plant pathogenic bacteria [J]. Sci Agric Sin, 2017, 50(12): 2305-2314. | |
| [52] | Adeolu M, Alnajar S, Naushad S, et al. Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov [J]. Int J Syst Evol Microbiol, 2016, 66(12): 5575-5599. |
| [1] | 沈川, 李夏, 覃剑锋, 段龙飞, 刘佳. 基于软腐病菌诱导的魔芋酵母双杂交文库筛选WRKY72互作蛋白[J]. 生物技术通报, 2025, 41(1): 85-94. |
| [2] | 马俊秀, 吴皓琼, 姜威, 闫更轩, 胡基华, 张淑梅. 蔬菜软腐病菌广谱拮抗细菌菌株筛选鉴定及防效研究[J]. 生物技术通报, 2023, 39(7): 228-240. |
| [3] | 文畅, 刘晨, 卢诗韵, 许忠兵, 艾超凡, 廖汉鹏, 周顺桂. 一株新的多重耐药福氏志贺菌噬菌体生物学特性及基因组分析[J]. 生物技术通报, 2022, 38(9): 127-135. |
| [4] | 李霁虹, 荆玉玲, 马桂珍, 郭荣君, 李世东. 无色杆菌77的基因组构成及其趋化和耐药特性[J]. 生物技术通报, 2022, 38(9): 136-146. |
| [5] | 刘警鞠, 张雨森, 陈娟, 孙炳达, 赵国柱. 曲霉属的现代分类命名研究进展[J]. 生物技术通报, 2022, 38(7): 109-118. |
| [6] | 韩东晶, 王志花, 周宁, 刘国庆, 杨少华, 汪文君. 白蚁菌圃中木质素降解菌的筛选及降解效果[J]. 生物技术通报, 2022, 38(3): 113-120. |
| [7] | 武利勤, 尚宏忠, 顾海科. 拮抗匍枝根霉的生防菌R1B的筛选鉴定和抑菌活析[J]. 生物技术通报, 2019, 35(4): 29-35. |
| [8] | 罗燕,刘小刚,周志钦. 植物糖基转移酶基因的分离方法及其生物学功能研究进展[J]. 生物技术通报, 2016, 32(12): 34-39. |
| [9] | 马宏;. 我国马铃薯软腐病防治的研究进展[J]. , 2007, 0(01): 42-44. |
| [10] | 孙国凤. 导入昆虫抗菌肽基因的烟草的开发[J]. , 1996, 0(01): 16-17. |
| [11] | 孙雷心. 生物技术:能救命干嘛不能进肚?[J]. , 1994, 0(01): 29-30. |
| [12] | 孙国凤;. 用细胞融合法育成了抗软腐病莴苣[J]. , 1988, 0(03): 13-14. |
| [13] | 孙国凤;. 麒麟啤酒公司再生马铃薯和番茄的融合植株成功[J]. , 1988, 0(03): 14-14. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||