







Biotechnology Bulletin ›› 2021, Vol. 37 ›› Issue (8): 307-318.doi: 10.13560/j.cnki.biotech.bull.1985.2021-0183
Previous Articles Next Articles
WANG Zhi-bo1,2( ), WANG Dao-ping2, MIAO Lan1,3, LI Ying1, PAN Ying-hong2(), LIU Jian-xun1,3()
), WANG Dao-ping2, MIAO Lan1,3, LI Ying1, PAN Ying-hong2(), LIU Jian-xun1,3()
Received:2021-02-09
Online:2021-08-26
Published:2021-09-10
Contact:
PAN Ying-hong,LIU Jian-xun
E-mail:wzb19950916@163.com;panyinghong@caas.cn;liujx0324@sina.com
WANG Zhi-bo, WANG Dao-ping, MIAO Lan, LI Ying, PAN Ying-hong, LIU Jian-xun. Comparative Study on Methods of Analyzing Proteome in Blood Samples[J]. Biotechnology Bulletin, 2021, 37(8): 307-318.
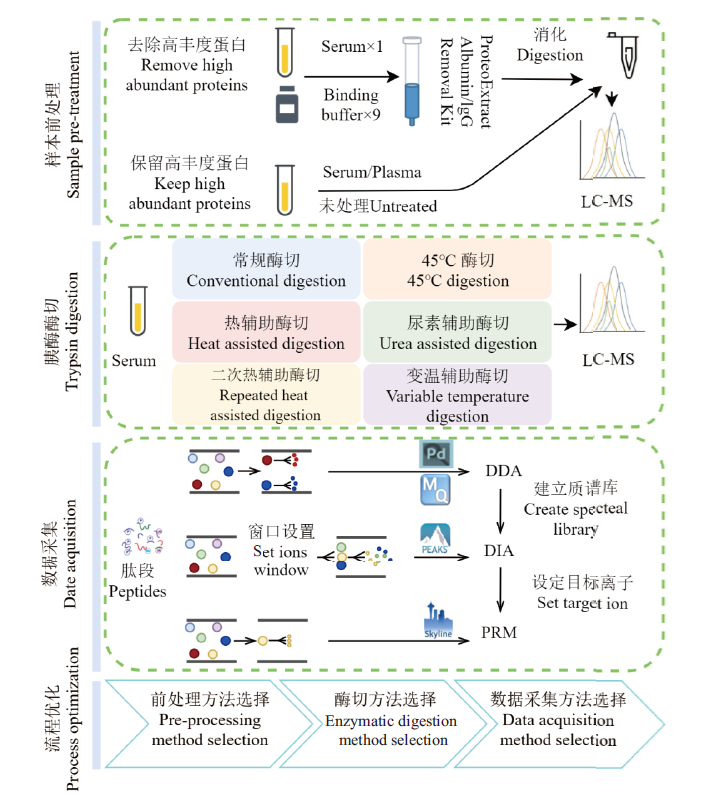
Fig.1 Experimental flow
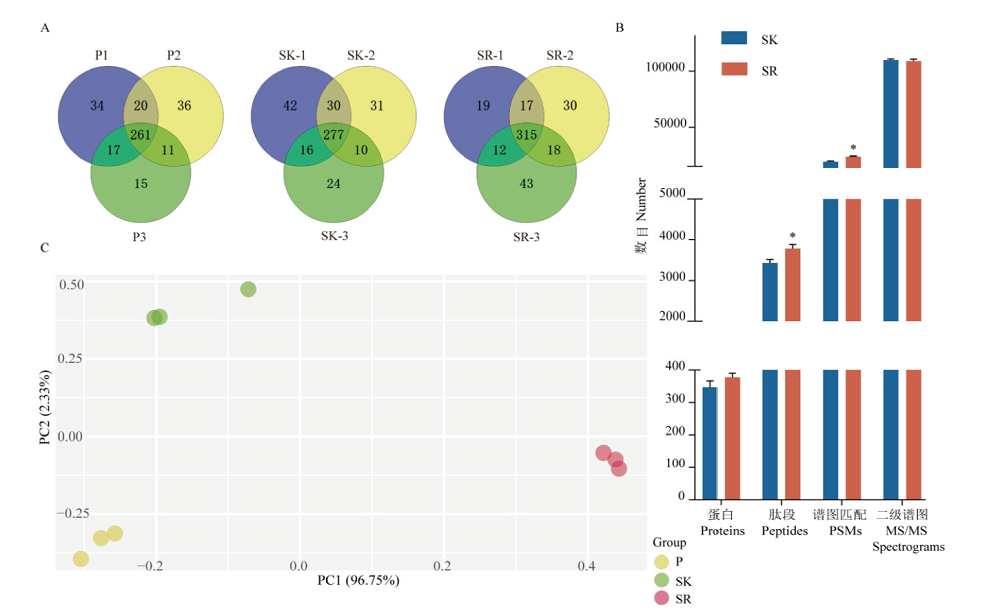
Fig. 2 Comparison of mass spectrometry results of blood samples prepared by 3 pre-processing methods A:Protein identification numbers and duplication rates of three repeated sample preparations for plasma(P),serum(SK)with high-abundance proteins remained,and serum(SR)with high-abundance proteins removed. B:The average number of proteins,peptides,PSMs and MS/MS spectrum for SK and SR. **:P<0.01. C:Principal component analysis of protein quantification

Fig. 3 Comparison of mass spectrometry results of serum proteins with different enzymatic digestion methods A:Average number of identified proteins,peptides and PSMs. *:P<0.05,and **:P<0.01. B:Comparison of percentage of missed enzymatic cleavage sites. C:Protein identification number and duplication rate of 3 repeated sample preparations. D:Sequence coverage of protein. E:Principal component analysis of protein quantification
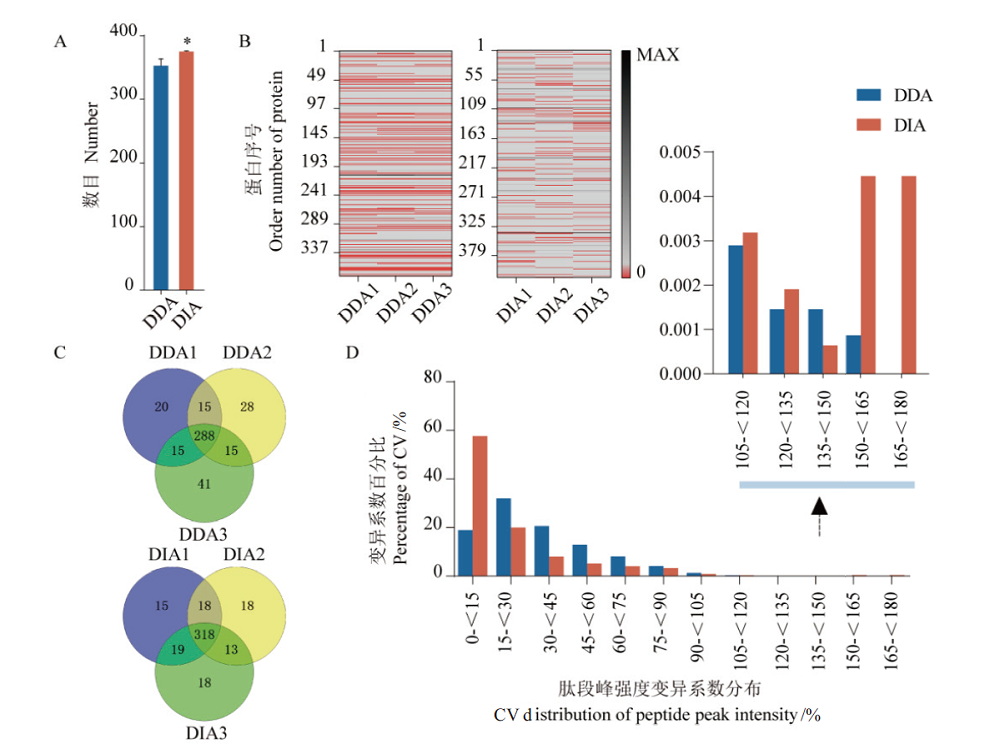
Fig. 4 Comparison of DDA and DIA data indices A:Average number of identified proteins in 3 technical repeats,*:P<0.05. B:Missing value matrix of protein quantification in 3 technical repeats,the red area represents missing values. C:Comparison of protein qualitative repeatability by 3 technical repeats. D:The statistics in coefficient of variation of the protein(peptides)peak intensity
| 蛋白 Protein | DIA* | PRM** |
|---|---|---|
| A0A0G2JSK1 | 0.163 | IFSQQADLSR,0.045;KIFSQQADLSR,0.064;DTLPHEDQGKGR,0.123 |
| A0A0G2JVP4 | 0.030 | ESATVTCLVK,0.058;TFPTLR,0.0732;DLPSPQK,0.128 |
| A0A0G2JV1 | 0.083 | HMEASLQEFKASPR,0.077;AVYLPNCDR,0.090;ISELKAEAVK,0.520 |
| A0A0G2K4K2 | 0.867 | LWIYDTSK,0.502 |
| A0A0G2K531 | 0.209 | QAALGAR,0.029;LFWEPMKIHDIR,0.460;NSCPPTAELLGSPGR,0.480 |
| A0A0G2K7X7 | 0.283 | CVGSAFETQSCNPER,0.019;ILPLTICK,0.019;ACGACPIWSK,0.032 |
| A0A0G2K896 | 0.217 | SSTFQLFGSPHGK,0.013;WCIVSDHEATK,0.059;VKWCAVGQQER,0.063 |
| B0BNN4 | 0.157 | SISCDEIPGQQSR,0.023;LCTPLLPK,0.031;HCYDIHNCVLK,0.129 |
| B2RYM3 | 0.052 | ELAAQTIK,0.024;ANLSSQILK,0.031;IADHKLSTFKADVR,0.049 |
| B5DEH7 | 0.114 | ANPGNFPWQAFTNIHGR,0.053;LPIADR,0.122;GLTVHLK,0.248 |
| D3ZAB3 | 0.711 | LMIYGATNLEDGVPSR,0.140 |
| D3ZAE6 | 1.732 | YLQGNTVQLR,0.085;QLDEGLFGR,0.450 |
| F1LWD0 | 0.867 | ANSYTTEYNPSVK,0.033 |
| F1LWS4 | 1.292 | NGYLYHENIRR,0.227;SYFPVPIGK,0.333;QCVFHYVENGESSYWQR,0.375 |
| F1LWW1 | 0.902 | VTISCR,1.485 |
| F1LXY6 | 0.040 | APEWLGFIR,0.037;ANGYTTEYNPSVK,0.040;AEDTATYYCAR,0.062 |
| F1LYU4 | 1.732 | ASNLASGIPAR,0.020 |
| G3V7L3 | 0.193 | TNVIQLR,0.019;CGTYGIYTK,0.066;LPITSLEK,0.080 |
| G3V7N9 | 0.055 | VITNVNDNYEPR,0.022;TVNSALRPNQAIR,0.057;TVNSALRPNQAIRFEK,0.146 |
| G3V7P2 | 0.250 | TLQEAVDSLKK,0.296 |
| G3V7P5 | 1.076 | WQSLPR,0.011;SCDVPVFENAK,0.022;IDHGSIKLPR,0.026 |
Table 1 Quantitative analysis on the coefficient of variation of protein from DIA and corresponding peptides from PRM in 3 repeats
| 蛋白 Protein | DIA* | PRM** |
|---|---|---|
| A0A0G2JSK1 | 0.163 | IFSQQADLSR,0.045;KIFSQQADLSR,0.064;DTLPHEDQGKGR,0.123 |
| A0A0G2JVP4 | 0.030 | ESATVTCLVK,0.058;TFPTLR,0.0732;DLPSPQK,0.128 |
| A0A0G2JV1 | 0.083 | HMEASLQEFKASPR,0.077;AVYLPNCDR,0.090;ISELKAEAVK,0.520 |
| A0A0G2K4K2 | 0.867 | LWIYDTSK,0.502 |
| A0A0G2K531 | 0.209 | QAALGAR,0.029;LFWEPMKIHDIR,0.460;NSCPPTAELLGSPGR,0.480 |
| A0A0G2K7X7 | 0.283 | CVGSAFETQSCNPER,0.019;ILPLTICK,0.019;ACGACPIWSK,0.032 |
| A0A0G2K896 | 0.217 | SSTFQLFGSPHGK,0.013;WCIVSDHEATK,0.059;VKWCAVGQQER,0.063 |
| B0BNN4 | 0.157 | SISCDEIPGQQSR,0.023;LCTPLLPK,0.031;HCYDIHNCVLK,0.129 |
| B2RYM3 | 0.052 | ELAAQTIK,0.024;ANLSSQILK,0.031;IADHKLSTFKADVR,0.049 |
| B5DEH7 | 0.114 | ANPGNFPWQAFTNIHGR,0.053;LPIADR,0.122;GLTVHLK,0.248 |
| D3ZAB3 | 0.711 | LMIYGATNLEDGVPSR,0.140 |
| D3ZAE6 | 1.732 | YLQGNTVQLR,0.085;QLDEGLFGR,0.450 |
| F1LWD0 | 0.867 | ANSYTTEYNPSVK,0.033 |
| F1LWS4 | 1.292 | NGYLYHENIRR,0.227;SYFPVPIGK,0.333;QCVFHYVENGESSYWQR,0.375 |
| F1LWW1 | 0.902 | VTISCR,1.485 |
| F1LXY6 | 0.040 | APEWLGFIR,0.037;ANGYTTEYNPSVK,0.040;AEDTATYYCAR,0.062 |
| F1LYU4 | 1.732 | ASNLASGIPAR,0.020 |
| G3V7L3 | 0.193 | TNVIQLR,0.019;CGTYGIYTK,0.066;LPITSLEK,0.080 |
| G3V7N9 | 0.055 | VITNVNDNYEPR,0.022;TVNSALRPNQAIR,0.057;TVNSALRPNQAIRFEK,0.146 |
| G3V7P2 | 0.250 | TLQEAVDSLKK,0.296 |
| G3V7P5 | 1.076 | WQSLPR,0.011;SCDVPVFENAK,0.022;IDHGSIKLPR,0.026 |
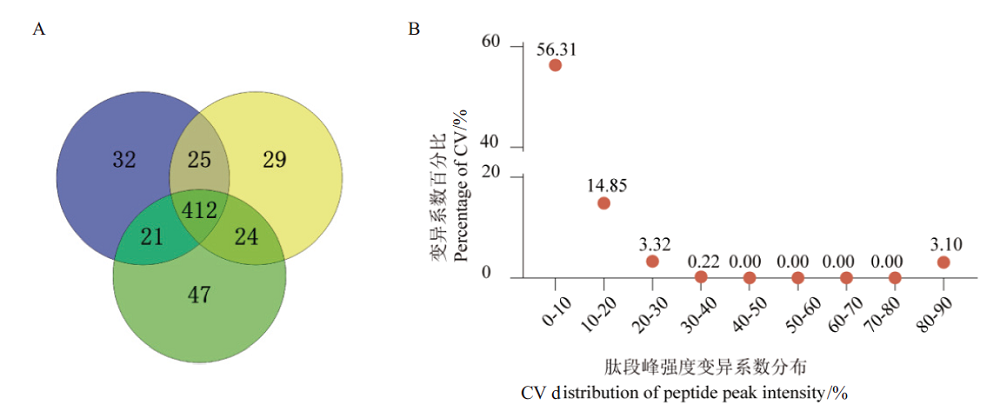
Fig.5 Qualitative and quantitative results of blood sample proteome based on optimized workflow A:Venn diagram of DDA qualitative results. B:CV distribution diagram of DDA protein quantitative results
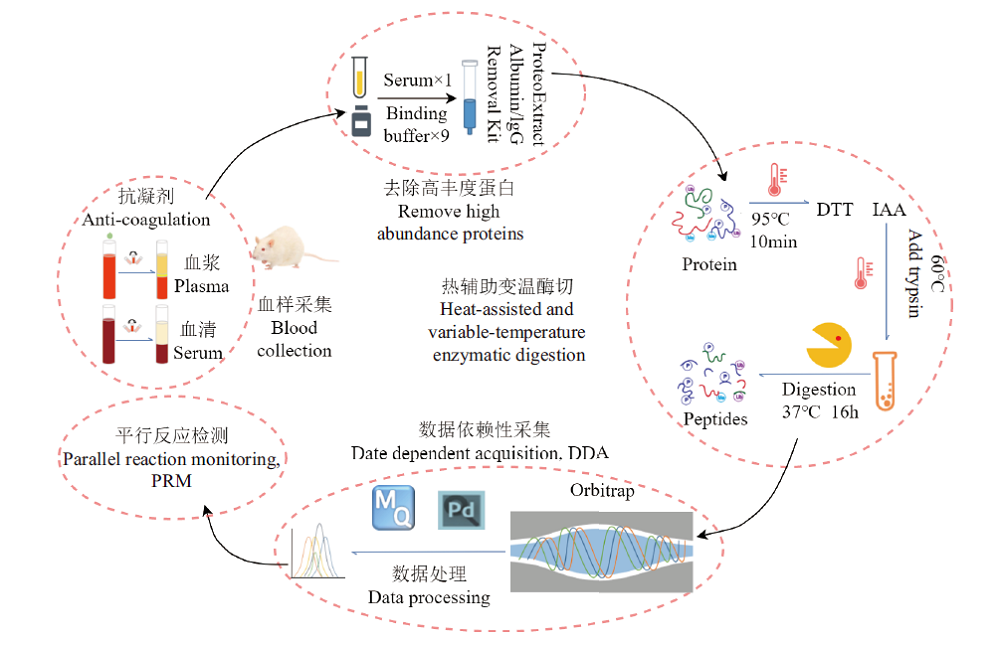
Fig. 6 Sample preparation and analysis workflow for blood proteome
| [1] |
Aslam B, Basit M, Nisar MA, et al. Proteomics:technologies and their applications[J]. J Chromatogr Sci, 2017, 55(2):182-196.
doi: 10.1093/chromsci/bmw167 URL |
| [2] |
Geyer PE, Holdt LM, Teupser D, et al. Revisiting biomarker discovery by plasma proteomics[J]. Mol Syst Biol, 2017, 13(9):942.
doi: 10.15252/msb.20156297 URL |
| [3] | Geyer PE, Kulak NA, Pichler G, et al. Plasma proteome profiling to assess human health and disease[J]. Cell Syst, 2016, 2(3):185-195. |
| [4] |
Peng L, Cantor DI, Huang C, et al. Tissue and plasma proteomics for early stage cancer detection[J]. Mol Omics, 2018, 14(6):405-423.
doi: 10.1039/C8MO00126J URL |
| [5] | 牟永莹, 王道平, 陈明, 等. 大豆种子蛋白质组样品制备与数据分析方法[J]. 生物技术通报, 2020, 36(12):247-255. |
| Mu YY, Wang DP, Chen M, et al. Sample preparation and data analysis method for soybean seed proteome[J]. Biotechnol Bull, 2020, 36(12):247-255. | |
| [6] | Wiśniewski JR. Filter-aided sample preparation:the versatile and efficient method for proteomic analysis[J]. Methods Enzymol, 2017, 585:15-27. |
| [7] |
Zolotarjova N, Martosella J, Nicol G, et al. Differences among techniques for high-abundant protein depletion[J]. Proteomics, 2005, 5(13):3304-3313.
pmid: 16052628 |
| [8] |
Polaskova V, Kapur A, Khan A, et al. High-abundance protein depletion:comparison of methods for human plasma biomarker discovery[J]. Electrophoresis, 2010, 31(3):471-482.
doi: 10.1002/elps.200900286 pmid: 20119956 |
| [9] |
Lima-Oliveira G, Monneret D, Guerber F, et al. Sample management for clinical biochemistry assays:Are serum and plasma interchangeable specimens?[J]. Crit Rev Clin Lab Sci, 2018, 55(7):480-500.
doi: 10.1080/10408363.2018.1499708 URL |
| [10] |
Park ZY, Russell DH. Thermal denaturation:a useful technique in peptide mass mapping[J]. Anal Chem, 2000, 72(11):2667-2670.
pmid: 10857653 |
| [11] |
Schniers A, Pasing Y, Hansen T. Lys-C/trypsin tandem-digestion protocol for gel-free proteomic analysis of colon biopsies[J]. Methods Mol Biol, 2019, 1959:113-122.
doi: 10.1007/978-1-4939-9164-8_7 pmid: 30852818 |
| [12] |
Betancourt LH, Sanchez A, Pla I, et al. Quantitative assessment of urea in-solution Lys-C/trypsin digestions reveals superior performance at room temperature over traditional proteolysis at 37℃[J]. J Proteome Res, 2018, 17(7):2556-2561.
doi: 10.1021/acs.jproteome.8b00228 pmid: 29812944 |
| [13] |
Budnik B, Levy E, Harmange G, et al. SCoPE-MS:mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation[J]. Genome Biol, 2018, 19(1):161.
doi: 10.1186/s13059-018-1547-5 URL |
| [14] |
Hu A, Noble WS, Wolf-Yadlin A. Technical advances in proteomics:new developments in data-independent acquisition[J]. F1000 Research, 2016, 5:419.
doi: 10.12688/f1000research URL |
| [15] |
Zhang Y, Fonslow BR, Shan B, et al. Protein analysis by shotgun/bottom-up proteomics[J]. Chem Rev, 2013, 113(4):2343-2394.
doi: 10.1021/cr3003533 URL |
| [16] |
Barkovits K, Pacharra S, Pfeiffer K, et al. Reproducibility, specificity and accuracy of relative quantification using spectral library-based data-independent acquisition[J]. Mol Cell Proteomics, 2020, 19(1):181-197.
doi: 10.1074/mcp.RA119.001714 pmid: 31699904 |
| [17] |
Venable JD, Dong MQ, Wohlschlegel J, et al. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra[J]. Nat Methods, 2004, 1(1):39-45.
pmid: 15782151 |
| [18] |
Bilbao A, Varesio E, Luban J, et al. Processing strategies and software solutions for data-independent acquisition in mass spectrometry[J]. Proteomics, 2015, 15(5/6):964-980.
doi: 10.1002/pmic.201400323 URL |
| [19] |
Shi T, Song E, Nie S, et al. Advances in targeted proteomics and applications to biomedical research[J]. Proteomics, 2016, 16(15/16):2160-2182.
doi: 10.1002/pmic.v16.15-16 URL |
| [20] |
Uzozie AC, Aebersold R. Advancing translational research and precision medicine with targeted proteomics[J]. J Proteomics, 2018, 189:1-10.
doi: 10.1016/j.jprot.2018.02.021 URL |
| [21] |
Silva ARM, Toyoshima MTK, Passarelli M, et al. Comparing data-independent acquisition and parallel reaction monitoring in their abilities to differentiate high-density lipoprotein subclasses[J]. J Proteome Res, 2020, 19(1):248-259.
doi: 10.1021/acs.jproteome.9b00511 URL |
| [22] |
Chiva C, Ortega M, Sabidó E. Influence of the digestion technique, protease, and missed cleavage peptides in protein quantitation[J]. J Proteome Res, 2014, 13(9):3979-3986.
doi: 10.1021/pr500294d URL |
| [23] |
Bruderer R, Muntel J, Müller S, et al. Analysis of 1508 plasma samples by capillary-flow data-independent acquisition profiles proteomics of weight loss and maintenance[J]. Mol Cell Proteomics, 2019, 18(6):1242-1254.
doi: 10.1074/mcp.RA118.001288 pmid: 30948622 |
| [24] |
Manes NP, Nita-Lazar A. Application of targeted mass spectrometry in bottom-up proteomics for systems biology research[J]. J Proteom, 2018, 189:75-90.
doi: 10.1016/j.jprot.2018.02.008 URL |
| [25] |
Woo CM, Lund PJ, Huang AC, et al. Mapping and quantification of over 2000 O-linked glycopeptides in activated human T cells with isotope-targeted glycoproteomics(isotag)[J]. Mol Cell Proteom, 2018, 17(4):764-775.
doi: 10.1074/mcp.RA117.000261 URL |
| [26] |
Qiu FH, Hou TY, Huang DH, et al. Evaluation of two high-abundance protein depletion kits and optimization of downstream isoelectric focusing[J]. Mol Med Rep, 2015, 12(5):7749-7755.
doi: 10.3892/mmr.2015.4417 URL |
| [27] |
Liu B, Qiu FH, Voss C, et al. Evaluation of three high abundance protein depletion kits for umbilical cord serum proteomics[J]. Proteome Sci, 2011, 9(1):24.
doi: 10.1186/1477-5956-9-24 URL |
| [28] |
Glatter T, Ludwig C, Ahrné E, et al. Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys-C/trypsin proteolysis over trypsin digestion[J]. J Proteome Res, 2012, 11(11):5145-5156.
doi: 10.1021/pr300273g pmid: 23017020 |
| [29] |
Kailasa S, Wu HF. Advances in nanomaterial-based microwaves and infrared wave-assisted tryptic digestion for ultrafast proteolysis and rapid detection by MALDI-MS[J]. Comb Chem High Throughput Screen, 2014, 17(1):68-79.
doi: 10.2174/1386207316666131110211353 URL |
| [1] | ZHOU Lu-qi, CUI Ting-ru, HAO Nan, ZHAO Yu-wei, ZHAO Bin, LIU Ying-chao. Application of Chemical Proteomics in Identifying the Molecular Targets of Natural Products [J]. Biotechnology Bulletin, 2023, 39(9): 12-26. |
| [2] | SANG Tian, WANG Peng-cheng. Research Progress in Plant SUMOylation [J]. Biotechnology Bulletin, 2023, 39(3): 1-12. |
| [3] | ZHAO Ming-ming, TANG Yin, GUO Lei-zhou, HAN Jia-hui, GE Jia-ming, MENG Yong, PING Shu-zhen, ZHOU Zheng-fu, WANG Jin. Function Analysis of Lon1 Protease Involved in High Temperature Stress and Cell Division of Deinococcus radiodurans R1 [J]. Biotechnology Bulletin, 2022, 38(5): 149-158. |
| [4] | LI Bing-juan, ZHENG Lu, SHEN Ren-fang, LAN Ping. Proteomic Analysis of RPP1A Involved in the Seedling Growth of Arabidopsis thaliana [J]. Biotechnology Bulletin, 2022, 38(2): 10-20. |
| [5] | MENG Li-ná, PENG Chun-ying, LI Tie-dong, LI Bo-sheng. Proteomic ánálysis of Spiruliná plátensis in Response to ársenic Stress [J]. Biotechnology Bulletin, 2020, 36(4): 107-116. |
| [6] | LI Kun, LIU Yue, HUANG Peng, YANG Zhi-fang, HU Qian, ZHANG Ying, LI Zhi-hong, LÜ Ye-hui, LIANG Le. Proteomics Study on Spermatogonia Differentiation in Mice [J]. Biotechnology Bulletin, 2020, 36(3): 168-176. |
| [7] | MU Yong-ying, WANG Dao-ping, CHEN Ming, QIU Li-juan, PAN Ying-hong. Sample Preparation and Data Analysis Method for Soybean Seed Proteome [J]. Biotechnology Bulletin, 2020, 36(12): 247-255. |
| [8] | ZHANG Liang, CHEN Xiao-qing, SONG Jia-yu, MAO Ran-ran, JIANG Qian-wen, LIN Xiang-min. Comparative Proteomics Analysis of Escherichia coli in Response to Barofloxacin Stress [J]. Biotechnology Bulletin, 2019, 35(3): 103-109. |
| [9] | LAN Yu-ting, WANG Shuang-Lei, LI Zheng-zhen, FENG Jin-chao, WANG Xiao-dong, SHI Sha. Research Advances in Proteomics of Ammopiptanthus in Responses to Abiotic Stresses [J]. Biotechnology Bulletin, 2019, 35(1): 112-119. |
| [10] | MU Yong-ying,GU Pei-ming,MA Bo,YAN Wen-xiu,WANG Dao-ping,PAN Ying-hong. Advancements in Quantitative Proteomics Technologies Based on Mass Spectrometry [J]. Biotechnology Bulletin, 2017, 33(9): 73-84. |
| [11] | SHAO Gui-fang, ZHANG Fan, WANG Jiao, ZHAO Kai, MO Yun-rong, DENG Ming-hua. Research Progress on Male Sterility of Pepper [J]. Biotechnology Bulletin, 2017, 33(8): 7-13. |
| [12] | ZHU Bei-bei, LI Xiang-yu, CHEN Huan, WANG Hong-juan, HOU Hong-wei, HU Qing-yuan. iTRAQ-based Quantitative Proteomic Analyses of Differentially Expressed Proteins in Nicotine-induced SH-SY5Y Cells [J]. Biotechnology Bulletin, 2017, 33(4): 90-97. |
| [13] | SONG Yan-chao, An Fei-fei, Xue Jing-jing, Qin Yu-ling, Li Kai-mian, CHEN Song-bi. Proteomic Analysis on Tuberous Roots of Cassava Cultivar ZM-Seaside and Mosaic-leaf Mutation [J]. Biotechnology Bulletin, 2017, 33(3): 78-85. |
| [14] | KONG De-kang, WANG Hong-qi, XU Jie, LIU Zi-li, WU Xiao-xiong. Applications of Genomics,Proteomics and Metabolomics in Microbial Degradation of PAHs [J]. Biotechnology Bulletin, 2017, 33(10): 46-51. |
| [15] | CHEN Fei, ZHOU Tong, WEI Yu-jia, YANG Jing, DAI Chuan-chao. Establishment of Membrane Proteomics Platform with Two-dimensional Electrophoresis for Preparing Identifying Plasma Membrane Proteins from Atractylodes lancea [J]. Biotechnology Bulletin, 2016, 32(9): 72-82. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||