







Biotechnology Bulletin ›› 2025, Vol. 41 ›› Issue (10): 143-155.doi: 10.13560/j.cnki.biotech.bull.1985.2025-0470
Previous Articles Next Articles
HE Yuan1,2( ), MOU Qiang1,2, HE Yu-bing2,3(), ZHAO Xiao-yan2, WANG Jian1,2, ZHOU Guo-min4,5,6, ZHANG Jian-hua1,2()
), MOU Qiang1,2, HE Yu-bing2,3(), ZHAO Xiao-yan2, WANG Jian1,2, ZHOU Guo-min4,5,6, ZHANG Jian-hua1,2()
Received:2025-05-08
Online:2025-10-26
Published:2025-10-28
Contact:
HE Yu-bing, ZHANG Jian-hua
E-mail:821012450699@caas.cn;heyubing@caas.cn;zhangjianhua@caas.cn
HE Yuan, MOU Qiang, HE Yu-bing, ZHAO Xiao-yan, WANG Jian, ZHOU Guo-min, ZHANG Jian-hua. Advances in Protein Mining and Design Based on Artificial Intelligence[J]. Biotechnology Bulletin, 2025, 41(10): 143-155.
工具名称 Tool name | 类型 Type | 功能概述 Function overview | 主要用途 Primary application |
|---|---|---|---|
| BLAST | 序列比对 | 通过序列相似性在数据库中查找相似蛋白 | 同源蛋白搜索、功能预测 |
| HMMER | 隐马尔可夫模型 | 用HMM识别蛋白家族保守序列模式 | 敏感的家族成员识别 |
| PSI-BLAST | 迭代序列比对 | 构建PSSM模型用于检测远缘同源 | 弱同源序列检测 |
| AlphaFold2 | 结构预测 | 高精度蛋白质三级结构建模 | 结构建模、结构基础的功能预测 |
| DALI | 结构比对 | 基于空间坐标的结构比对与聚类 | 蛋白质结构相似性分析 |
| TM-align | 结构比对 | 比较两个结构的拓扑关系,输出TM-score | 判断是否具有相似折叠模式 |
| InterProScan | 结构域识别工具 | 整合多个数据库识别功能结构域 | 蛋白注释、家族归类 |
| EFI-EST | 序列相似性网络 | 构建Enzyme Similarity Network(ESN)图 | 可视化蛋白功能空间、聚类分析 |
| DeepFRI | 深度学习 | 基于结构/序列的功能预测 | AI辅助的功能预测 |
Table 1 Major tools for protein mining
工具名称 Tool name | 类型 Type | 功能概述 Function overview | 主要用途 Primary application |
|---|---|---|---|
| BLAST | 序列比对 | 通过序列相似性在数据库中查找相似蛋白 | 同源蛋白搜索、功能预测 |
| HMMER | 隐马尔可夫模型 | 用HMM识别蛋白家族保守序列模式 | 敏感的家族成员识别 |
| PSI-BLAST | 迭代序列比对 | 构建PSSM模型用于检测远缘同源 | 弱同源序列检测 |
| AlphaFold2 | 结构预测 | 高精度蛋白质三级结构建模 | 结构建模、结构基础的功能预测 |
| DALI | 结构比对 | 基于空间坐标的结构比对与聚类 | 蛋白质结构相似性分析 |
| TM-align | 结构比对 | 比较两个结构的拓扑关系,输出TM-score | 判断是否具有相似折叠模式 |
| InterProScan | 结构域识别工具 | 整合多个数据库识别功能结构域 | 蛋白注释、家族归类 |
| EFI-EST | 序列相似性网络 | 构建Enzyme Similarity Network(ESN)图 | 可视化蛋白功能空间、聚类分析 |
| DeepFRI | 深度学习 | 基于结构/序列的功能预测 | AI辅助的功能预测 |
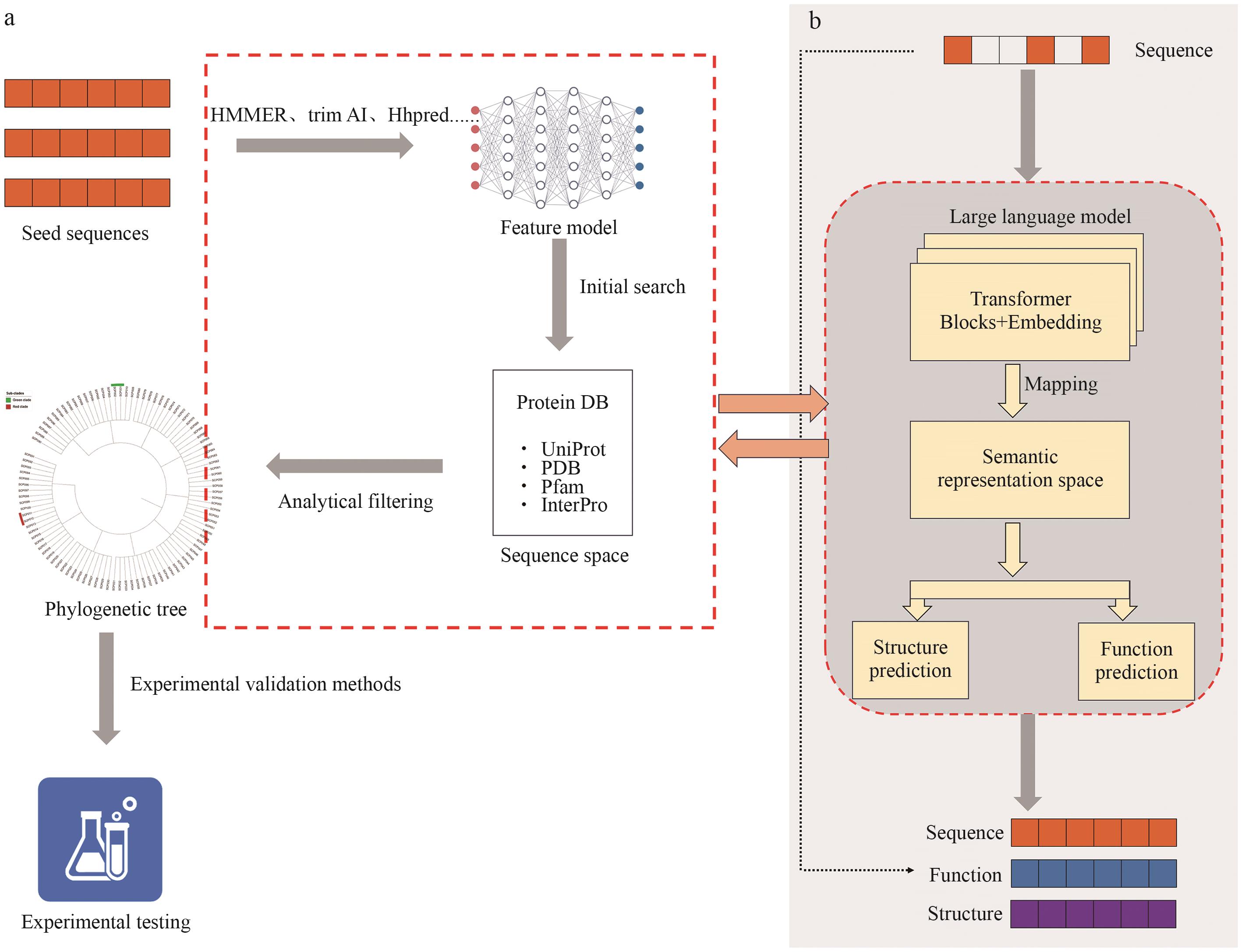
Fig. 1 Comparison of sequence similarity-based protein mining approachesa: Representative seed sequences are selected from databases and used for homology search and feature extraction via tools such as HMMER and HHpred, forming an initial candidate space. Phylogenetic analysis and functional annotations are then applied for further filtering, followed by experimental validation of potential functions. b: Raw amino acid sequences are used as input for large language models (e.g., Transformer-based architectures) to learn latent semantic representations, enabling structure and function prediction tasks and yielding candidate sequences, structures, and functional labels
比较维度 Comparison dimension | 序列相似性方法 Sequence similarity-based methods | 结构相似性方法 Structure similarity-based methods |
|---|---|---|
| 核心依据 | 氨基酸序列保守性 | 三维结构的空间拓扑与折叠方式 |
| 代表工具 | BLAST, HMMER, PSI-BLAST | DALI, TM-align, Foldseek |
| 检测远缘关系能力 | 中等(需要某些序列保守性) | 强(可发现序列不同但结构相似的蛋白) |
| 数据需求 | 仅需序列信息 | 需要可靠的结构信息(如PDB或AlphaFold预测) |
| 运算复杂度 | 相对较低 | 较高(结构比对更耗时) |
| 适用场景 | 大规模快速筛查、同源注释 | 深层功能预测、新折叠模式发现 |
| 局限性 | 对进化遥远蛋白不敏感 | 受限于结构预测精度、运算资源 |
Table 2 Comparison between sequence similarity-based and structure similarity-based protein mining
比较维度 Comparison dimension | 序列相似性方法 Sequence similarity-based methods | 结构相似性方法 Structure similarity-based methods |
|---|---|---|
| 核心依据 | 氨基酸序列保守性 | 三维结构的空间拓扑与折叠方式 |
| 代表工具 | BLAST, HMMER, PSI-BLAST | DALI, TM-align, Foldseek |
| 检测远缘关系能力 | 中等(需要某些序列保守性) | 强(可发现序列不同但结构相似的蛋白) |
| 数据需求 | 仅需序列信息 | 需要可靠的结构信息(如PDB或AlphaFold预测) |
| 运算复杂度 | 相对较低 | 较高(结构比对更耗时) |
| 适用场景 | 大规模快速筛查、同源注释 | 深层功能预测、新折叠模式发现 |
| 局限性 | 对进化遥远蛋白不敏感 | 受限于结构预测精度、运算资源 |
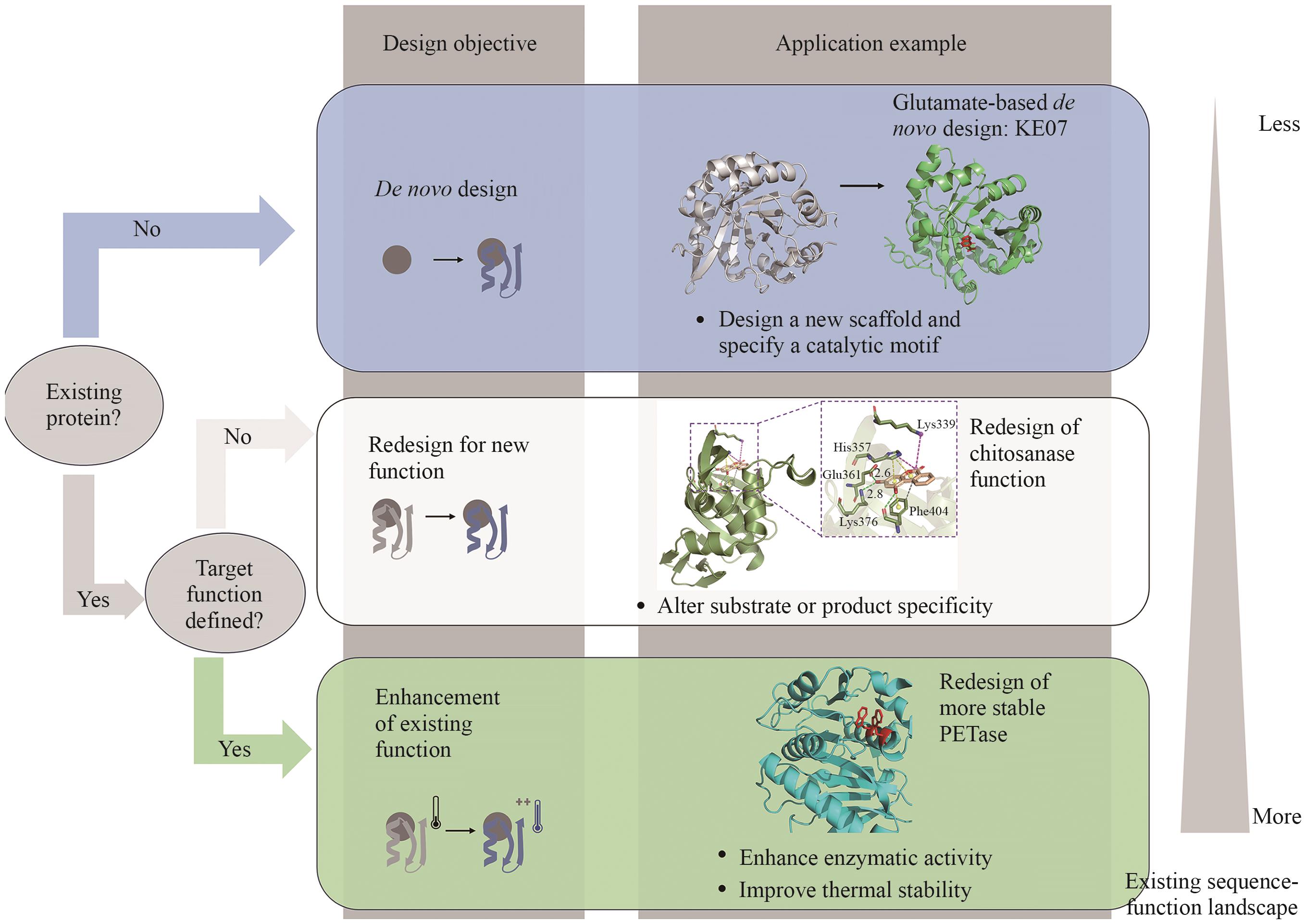
Fig. 2 Design objectives of proteins
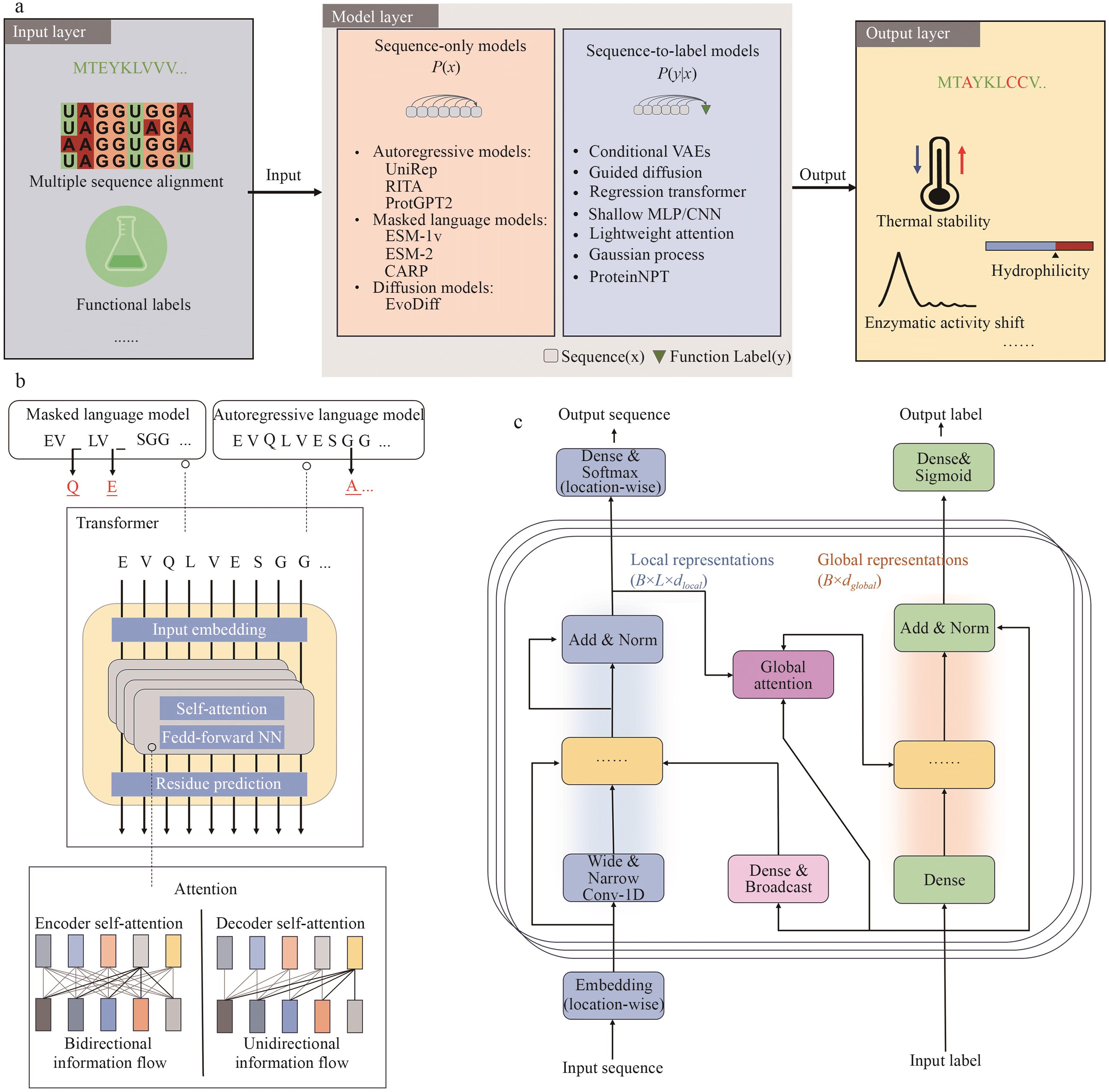
Fig. 3 Sequence-based protein designa: The input layer includes multiple sequence alignment (MSA) data and functional labels. The model layer is divided into sequence-only models and sequence-to-label models. The output layer yields predicted target sequences or functional properties, such as thermal stability, solubility, or changes in catalytic activity. b: The upper panel illustrates the difference between masked language models and autoregressive language models. The lower panel shows the typical transformer architecture, including input embedding, attention mechanisms, and residual connections. Masked models support bidirectional information flow for contextual modeling, while autoregressive models operate with unidirectional flow for sequence generation. c: Given protein sequences and functional labels as inputs, the model encodes local features through convolutional layers and global context via attention mechanisms. These representations are then fused to generate target sequences or predict functional labels. The output can be modeled using a softmax layer for sequence generation or a sigmoid layer for multi-label classification
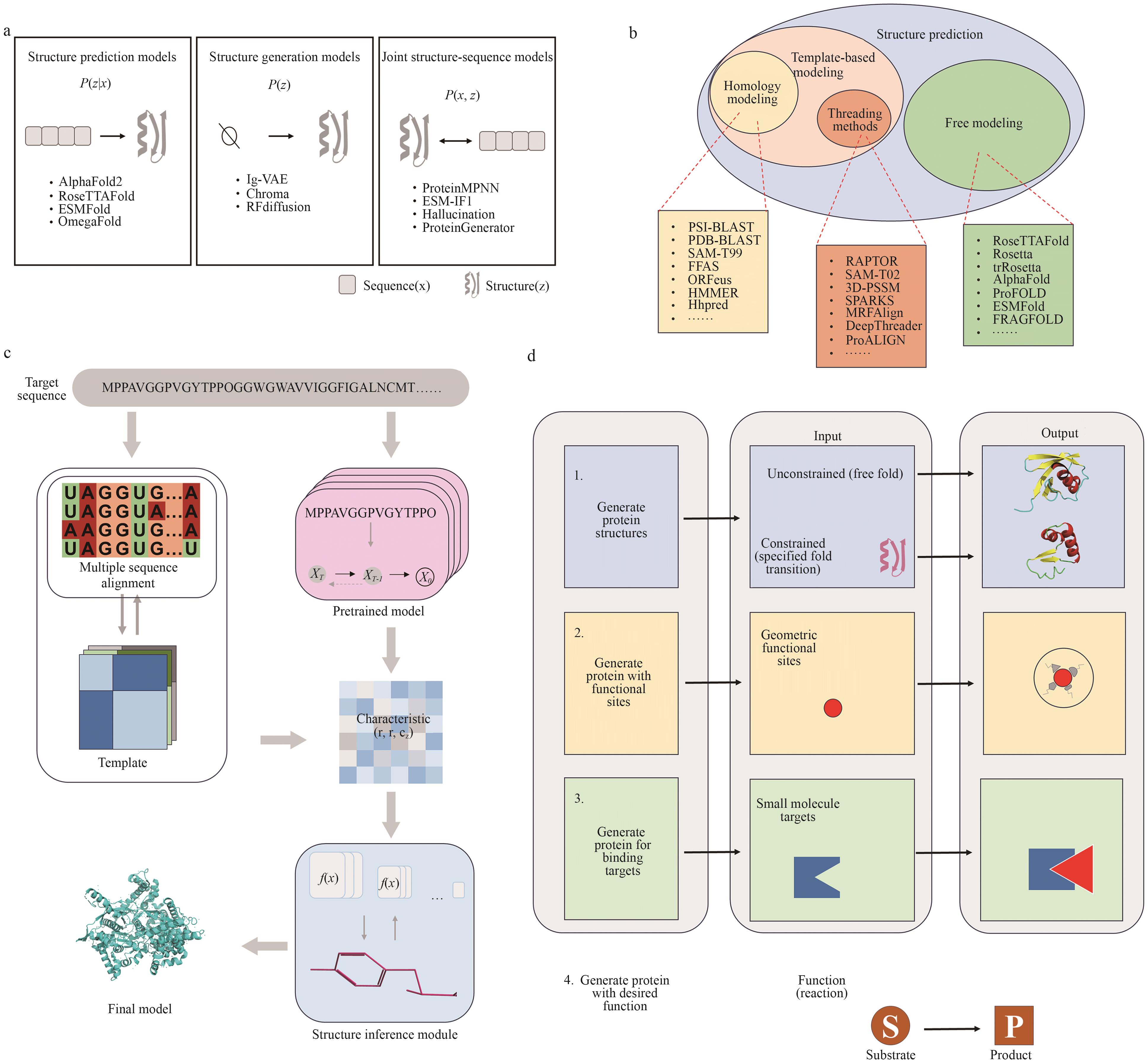
Fig. 4 Structure-based protein designa: Structure prediction models aim to infer protein structures from sequences by learning P(xz); structure generation models directly model the structural distribution P(z); and joint structure-sequence models learn the joint distributionP(x, z), enabling coordinated sequence-structure design. b: Template-based methods, including homology modeling and threading, rely on known protein structure templates, while free modeling methods generate structures without using templates. The figure categorizes representative structure prediction tools according to their methodological classes. c: Template information is generated via multiple sequence alignment and fed into pretrained models to extract residue-residue features. These are combined with structural inference modules to predict atomic coordinates, enabling high-accuracy structure modeling. This workflow integrates evolutionary information with geometric learning.d: Structure generation can be categorized into four stages based on functional objectives: (1) generating proteins with properly folded structures; (2) constructing proteins with specified geometric functional sites; (3) enhancing specific binding affinity to small molecules; and (4) designing proteins with catalytic functions that enable the transformation of substrate S into product P
| [1] | Analytical Methods Committee, AMCTB No 59. PCR-the polymerase chain reaction [J]. Anal Methods, 2014, 6(2): 333-336. |
| [2] | Chen K, Arnold FH. Tuning the activity of an enzyme for unusual environments: sequential random mutagenesis of subtilisin E for catalysis in dimethylformamide [J]. Proc Natl Acad Sci USA, 1993, 90(12): 5618-5622. |
| [3] | Samish I. Achievements and challenges in computational protein design [J]. Methods Mol Biol, 2017, 1529: 21-94. |
| [4] | Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold [J]. Nature, 2021, 596(7873): 583-589. |
| [5] | Evans R, O’Neill M, Pritzel A, et al. Protein complex prediction with AlphaFold-Multimer [J]. bioRxiv, 2022. DOI: https://doi.org/10.1101/2021.10.04.463034 . |
| [6] | Baek M, McHugh R, Anishchenko I, et al. Accurate prediction of protein-nucleic acid complexes using RoseTTAFoldNA [J]. Nat Methods, 2024, 21(1): 117-121. |
| [7] | Elnaggar A, Heinzinger M, Dallago C, et al. ProtTrans: toward understanding the language of life through self-supervised learning [J]. IEEE Trans Pattern Anal Mach Intell, 2022, 44(10): 7112-7127. |
| [8] | Gligorijević V, Renfrew PD, Kosciolek T, et al. Structure-based protein function prediction using graph convolutional networks [J]. Nat Commun, 2021, 12: 3168. |
| [9] | FerruzN, SchmidtS, HöckerB. ProtGPT2 is a deep unsupervised language model for protein design [J]. Nat Commun, 2022, 13(1): 4348. |
| [10] | Wang CK, Wang CP, Wang Z, et al. DeepDirect: learning directions of social ties with edge-based network embedding [J]. IEEE Trans Knowl Data Eng, 2019, 31(12): 2277-2291. |
| [11] | Watson JL, Juergens D, Bennett NR, et al. De novo design of protein structure and function with RFdiffusion [J]. Nature, 2023, 620(7976): 1089-1100. |
| [12] | Gligorijević V, Renfrew PD, Kosciolek T, et al. Structure-based protein function prediction using graph convolutional networks [J]. Nat Commun, 2021, 12(1): 3168. |
| [13] | Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3 [J]. Nature, 2024, 630(8016): 493-500. |
| [14] | 池燕飞, 李春, 冯旭东. 机器学习在蛋白质功能预测领域的研究进展 [J]. 生物工程学报, 2023, 39(6): 2141-2157. |
| Chi YF, Li C, Feng XD. Advances in machine learning for predicting protein functions [J]. Chin J Biotechnol, 2023, 39(6): 2141-2157. | |
| [15] | Xu K, Feng H, Zhang HH, et al. Structure-guided discovery of highly efficient cytidine deaminases with sequence-context independence [J]. Nat Biomed Eng, 2025, 9(1): 93-108. |
| [16] | Jiang KY, Lim J, Sgrizzi S, et al. Programmable RNA-guided DNA endonucleases are widespread in eukaryotes and their viruses [J]. Sci Adv, 2023, 9(39): eadk0171. |
| [17] | Wang XR, Yin XD, Jiang DJ, et al. Multi-modal deep learning enables efficient and accurate annotation of enzymatic active sites [J]. Nat Commun, 2024, 15(1): 7348. |
| [18] | Kucera T, Togninalli M, Meng-Papaxanthos L. Conditional generative modeling for de novo protein design with hierarchical functions [J]. Bioinformatics, 2022, 38(13): 3454-3461. |
| [19] | Kulmanov M, Khan MA, Hoehndorf R, et al. DeepGO: predicting protein functions from sequence and interactions using a deep ontology-aware classifier [J]. Bioinformatics, 2018, 34(4): 660-668. |
| [20] | Holehouse AS, Kragelund BB. The molecular basis for cellular function of intrinsically disordered protein regions [J]. Nat Rev Mol Cell Biol, 2024, 25(3): 187-211. |
| [21] | Mukhopadhyay R, Irausquin S, Schmidt C, et al. Dynafold: a dynamic programming approach to protein backbone structure determination from minimal sets of Residual Dipolar Couplings [J]. J Bioinform Comput Biol, 2014, 12(1): 1450002. |
| [22] | Wang T, He XH, Li MY, et al. Ab initio characterization of protein molecular dynamics with AI2BMD [J]. Nature, 2024, 635(8040): 1019-1027. |
| [23] | Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality [J]. Proteins, 2004, 57(4): 702-710. |
| [24] | Hou MH, Xia Y, Wang PC, et al. High-accuracy protein complex structure modeling based on sequence-derived structure complementarity [J]. bioRxiv, 2025: 2025.03.26.645390. |
| [25] | Peng CX, Zhou XG, Liu J, et al. Multiple conformational states assembly of multidomain proteins using evolutionary algorithm based on structural analogues and sequential homologues [J]. Fundam Res, 2024, 6(4): 223-236. |
| [26] | Shaw DE, Maragakis P, Lindorff-Larsen K, et al. Atomic-level characterization of the structural dynamics of proteins [J]. Science, 2010, 330(6002): 341-346. |
| [27] | Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool [J]. J Mol Biol, 1990, 215(3): 403-410. |
| [28] | Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching [J]. Nucleic Acids Res, 2011, 39(Web Server issue): W29-W37. |
| [29] | Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs [J]. Nucleic Acids Res, 1997, 25(17): 3389-3402. |
| [30] | van Kempen M, Kim SS, Tumescheit C, et al. Fast and accurate protein structure search with Foldseek [J]. Nat Biotechnol, 2024, 42(2): 243-246. |
| [31] | Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score [J]. Nucleic Acids Res, 2005, 33(7): 2302-2309. |
| [32] | Dauparas J, Anishchenko I, Bennett N, et al. Robust deep learning-based protein sequence design using ProteinMPNN [J]. Science, 2022, 378(6615): 49-56. |
| [33] | 操帆, 陈耀晞, 缪阳洋, 等. 蛋白质计算设计: 方法和应用展望 [J]. 合成生物学, 2021, 2(1): 15-32. |
| Cao F, Chen YX, Miao YY, et al. Computational protein design: perspectives in methods and applications [J]. Synth Biol J, 2021, 2(1): 15-32. | |
| [34] | Notin P, Rollins N, Gal Y, et al. Machine learning for functional protein design [J]. Nat Biotechnol, 2024, 42(2): 216-228. |
| [35] | Lu HY, Diaz DJ, Czarnecki NJ, et al. Machine learning-aided engineering of hydrolases for PET depolymerization [J]. Nature, 2022, 604(7907): 662-667. |
| [36] | Sumida KH, Núñez-Franco R, Kalvet I, et al. Improving protein expression, stability, and function with ProteinMPNN [J]. J Am Chem Soc, 2024, 146(3): 2054-2061. |
| [37] | Gercke D, Regel EK, Singh R, et al. Rational protein design of Bacillus sp. MN chitosanase for altered substrate binding and production of specific chitosan oligomers [J]. J Biol Eng, 2019, 13: 23. |
| [38] | Khersonsky O, Röthlisberger D, Dym O, et al. Evolutionary optimization of computationally designed enzymes: kemp eliminases of the KE07 series [J]. J Mol Biol, 2010, 396(4): 1025-1042. |
| [39] | Tian XC, Wang ZY, Yang KK, et al. Sequence vs. structure: delving deep into data-driven protein function prediction [J]. bioRxiv, 2023. DOI: https://doi.org/10.1101/2023.04.02.534383 . |
| [40] | 刘南, 金小程, 杨崇周, 等. 人工智能时代下的蛋白质从头设计 [J]. 生物工程学报, 2024, 40(11): 3912-3929. |
| Liu N, Jin XC, Yang CZ, et al. De novo protein design in the era of artificial intelligence [J]. Chin J Biotechnol, 2024, 40(11): 3912-3929. | |
| [41] | 康里奇, 谈攀, 洪亮. 人工智能时代下的酶工程 [J]. 合成生物学, 2023, 4(3): 524-534. |
| Kang LQ, Tan P, Hong L. Enzyme engineering in the age of artificial intelligence [J]. Synth Biol J, 2023, 4(3): 524-534. | |
| [42] | Kasahara K, Terazawa H, Takahashi T, et al. Studies on molecular dynamics of intrinsically disordered proteins and their fuzzy complexes: a mini-review [J]. Comput Struct Biotechnol J, 2019, 17: 712-720. |
| [43] | Lin ZM, Akin H, Rao R, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model [J]. Science, 2023, 379(6637): 1123-1130. |
| [44] | Madani A, Krause B, Greene ER, et al. Large language models generate functional protein sequences across diverse families [J]. Nat Biotechnol, 2023, 41(8): 1099-1106. |
| [45] | Du ZY, Su H, Wang WK, et al. The trRosetta server for fast and accurate protein structure prediction [J]. Nat Protoc, 2021, 16(12): 5634-5651. |
| [46] | Bepler T, Berger B. Learning the protein language: Evolution, structure, and function [J]. Cell Syst, 2021, 12(6): 654-669.e3. |
| [47] | Ruffolo JA, Madani A. Designing proteins with language models [J]. Nat Biotechnol, 2024, 42(2): 200-202. |
| [48] | Vaswani A, Shazeer N, Parmar N, et al. Attention is all you need [J]. Adv Neural Inf Process Syst, 2017, 30: 5998-6008. |
| [49] | Shin JE, Riesselman AJ, Kollasch AW, et al. Protein design and variant prediction using autoregressive generative models [J]. Nat Commun, 2021, 12(1): 2403. |
| [50] | Alley EC, Khimulya G, Biswas S, et al. Unified rational protein engineering with sequence-based deep representation learning [J]. Nat Methods, 2019, 16(12): 1315-1322. |
| [51] | Brandes N, Ofer D, Peleg Y, et al. ProteinBERT: a universal deep-learning model of protein sequence and function [J]. Bioinformatics, 2022, 38(8): 2102-2110. |
| [52] | Liu X, Zhang FJ, Hou ZY, et al. Self-supervised learning: generative or contrastive [J]. IEEE Trans Knowl Data Eng, 2021, 35(1): 857-876. |
| [53] | Biswas S, Khimulya G, Alley EC, et al. Low-N protein engineering with data-efficient deep learning [J]. Nat Methods, 2021, 18(4): 389-396. |
| [54] | Yang RZ, Sun XY, Narasimhan K. A generalized algorithm for multi-objective reinforcement learning and policy adaptation [J]. CoRR., 2019. DOI: 10.48550/arxiv.1908.08342 |
| [55] | Karimi M, Zhu SW, Cao Y, et al. De novo protein design for novel folds using guided conditional Wasserstein generative adversarial networks [J]. J Chem Inf Model, 2020, 60(12): 5667-5681. |
| [56] | Hawkins-Hooker A, Depardieu F, Baur S, et al. Generating functional protein variants with variational autoencoders [J]. PLoS Comput Biol, 2021, 17(2): e1008736. |
| [57] | Noor MS, Ferdous S, Salehi R, et al. Next-generation metabolic models informed by biomolecular simulations [J]. Curr Opin Biotechnol, 2025, 92: 103259. |
| [58] | Notin P, Marks DS, Weitzman R, et al. ProteinNPT: improving protein property prediction and design with non-parametric transformers [J]. bioRxiv, 2023: 2023.12.06.570473. |
| [59] | Branden CI, Tooze J. Introduction to protein structure [M]. New York: Garland Science, 2012. |
| [60] | Dill KA, MacCallum JL. The protein-folding problem, 50 years on [J]. Science, 2012, 338(6110): 1042-1046. |
| [61] | Yang JY, Yan RX, Roy A, et al. The I-TASSER suite: protein structure and function prediction [J]. Nat Methods, 2015, 12(1): 7-8. |
| [62] | Kuhlman B, Bradley P. Advances in protein structure prediction and design [J]. Nat Rev Mol Cell Biol, 2019, 20(11): 681-697. |
| [63] | Anfinsen CB. Principles that govern the folding of protein chains [J]. Science, 1973, 181(4096): 223-230. |
| [64] | Huang B, Kong LP, Wang C, et al. Protein structure prediction: challenges, advances, and the shift of research paradigms [J]. Genom Proteom Bioinform, 2023, 21(5): 913-925. |
| [65] | Peng CX, Liang F, Xia YH, et al. Recent advances and challenges in protein structure prediction [J]. J Chem Inf Model, 2024, 64(1): 76-95. |
| [66] | Wang S, Sun SQ, Li Z, et al. Accurate de novo prediction of protein contact map by ultra-deep learning model [J]. PLoS Comput Biol, 2017, 13(1): e1005324. |
| [67] | Ingraham J, Riesselman A, Sander C, Marks D. Learning protein structure with a differentiable simulator [C]. Proceedings of the International Conference on Learning Representations, 2019. |
| [68] | 陈志航, 季梦麟, 戚逸飞. 人工智能蛋白质结构设计算法研究进展 [J]. 合成生物学, 2023, 4(3): 464-487. |
| Chen ZH, Ji ML, Qi YF. Research progress on artificial intelligence-based protein structure design algorithms [J]. Synth Biol J, 2023, 4(3): 464-487. | |
| [69] | Liu Y, Kuhlman B. RosettaDesign server for protein design [J]. Nucleic Acids Res, 2006, 34(Web Server issue): W235-W238. |
| [70] | Hopf TA, Green AG, Schubert B, et al. The EVcouplings Python framework for coevolutionary sequence analysis [J]. Bioinformatics, 2019, 35(9): 1582-1584. |
| [71] | Lisanza SL, Gershon JM, Tipps SWK, et al. Multistate and functional protein design using RoseTTAFold sequence space diffusion [J]. Nat Biotechnol, 2025, 43(8): 1288-1298. |
| [72] | Wu KE, Yang KK, van den Berg R, et al. Protein structure generation via folding diffusion [J]. Nat Commun, 2024, 15(1): 1059. |
| [73] | Ingraham JB, Baranov M, Costello Z, et al. Illuminating protein space with a programmable generative model [J]. Nature, 2023, 623(7989): 1070-1078. |
| [1] | CAI Ru-feng, YANG Yu-xuan, YU Ji-zheng, LI Jia-nan. Artificial Intelligence Transforms Protein Engineering: From Structural Analysis to Synthetic Biology through Algorithmic Advancements [J]. Biotechnology Bulletin, 2025, 41(8): 1-10. |
| [2] | WANG Hui, FAN Ling-xi, SUN Ji-lu, WANG Yuan, WU Ning-feng, TIAN Jian, GUAN Fei-fei. Enhancing the Thermostability of Lysozyme RPL187 Based on Protein Intelligence Models [J]. Biotechnology Bulletin, 2025, 41(7): 336-346. |
| [3] | JI Hong-chao, LI Zheng-yan. Research Progress and Prospects in the Structural Annotation of Unknown Secondary Metabolites Based on Mass Spectrometry [J]. Biotechnology Bulletin, 2024, 40(10): 76-85. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||