







生物技术通报 ›› 2025, Vol. 41 ›› Issue (2): 97-106.doi: 10.13560/j.cnki.biotech.bull.1985.2024-0358
宋英培( ), 王灿, 周会汶, 孔可可, 许孟歌, 王瑞凯()
), 王灿, 周会汶, 孔可可, 许孟歌, 王瑞凯()
收稿日期:2024-04-14
出版日期:2025-02-26
发布日期:2025-02-28
通讯作者:
王瑞凯,男,博士,副教授,研究方向 :大豆遗传育种;E-mail: rikswang@sina.com作者简介:宋英培,女,博士,副教授,研究方向 :大豆遗传育种;E-mail: jiatai_105@163.com
基金资助:
SONG Ying-pei(), WANG Can, ZHOU Hui-wen, KONG Ke-ke, XU Meng-ge, WANG Rui-kai()
Received:2024-04-14
Published:2025-02-26
Online:2025-02-28
摘要:
目的 探究抗裂荚分子机制,挖掘抗裂荚大豆种质资源,可为揭示大豆裂荚性遗传和驯化机理、加速南方大豆抗裂荚新品种的选育提供依据。 方法 结合全基因组关联分析定位和野生栽培大豆群体遗传多样性分析,探寻大豆裂荚性相关基因。使用302份大豆材料进行2年表型测定,借助95 744个单核苷酸多态性标记进行全基因组关联分析。使用包括1 308份栽培大豆和203份野生大豆的重测序数据进行QTL区段序列的遗传多样性分析。 结果 通过全基因组关联分析检测到3个表型变异解释率大于10%的QTL位点,分别为qPdh-Chr08(Gm08:3048312)、qPdh-Chr15(Gm15:312814)和qPdh-Chr16(Gm16:29951529)。其中qPdh-Chr16是已知基因pdh1(Pod dehiscence habit 1)。结合野生和栽培大豆重测序数据发现,qPdh-Chr08和qPdh-Chr15内存在人工选择导致的群体分化序列,发现2个重要候选基因Glyma.08G038600和Glyma.15G003600。根据功能注释发现,这两个基因参与生长素和木质素的代谢。 结论 使用2年数据检测到大豆裂荚性2个新的QTL位点qPdh-Chr08和qPdh-Chr15,并在QTL区域内鉴定野生栽培大豆分化基因,最终筛选到2个重要候选基因。
宋英培, 王灿, 周会汶, 孔可可, 许孟歌, 王瑞凯. 基于全基因组关联分析和遗传多样性的大豆裂荚性状解析[J]. 生物技术通报, 2025, 41(2): 97-106.
SONG Ying-pei, WANG Can, ZHOU Hui-wen, KONG Ke-ke, XU Meng-ge, WANG Rui-kai. Analysis of Soybean Pod Dehiscence Habit Based on Whole Genome Association Analysis and Genetic Diversity[J]. Biotechnology Bulletin, 2025, 41(2): 97-106.
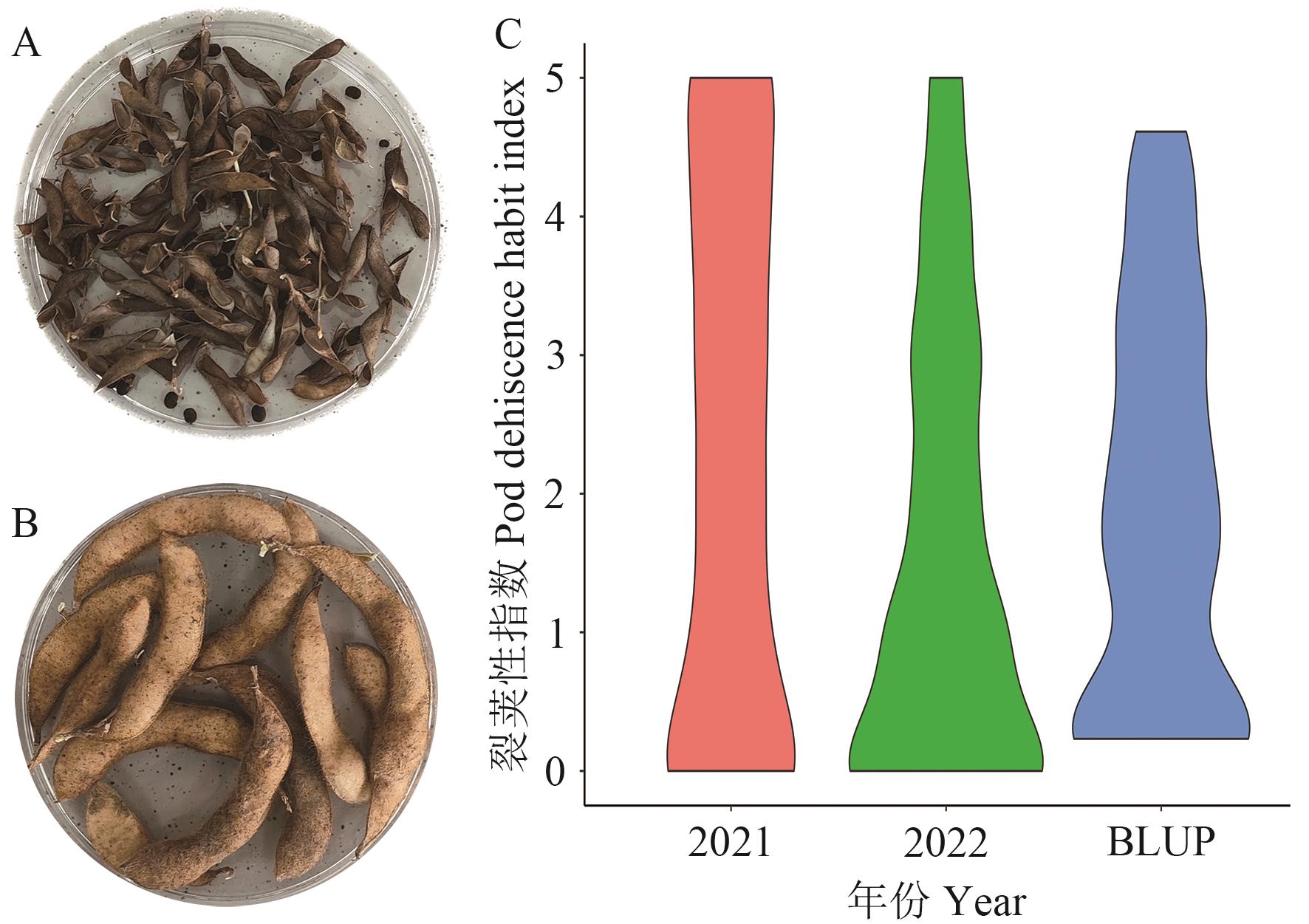
图1 裂荚性表型分布
Fig. 1 Phenotypic distribution of pod dehiscence habit
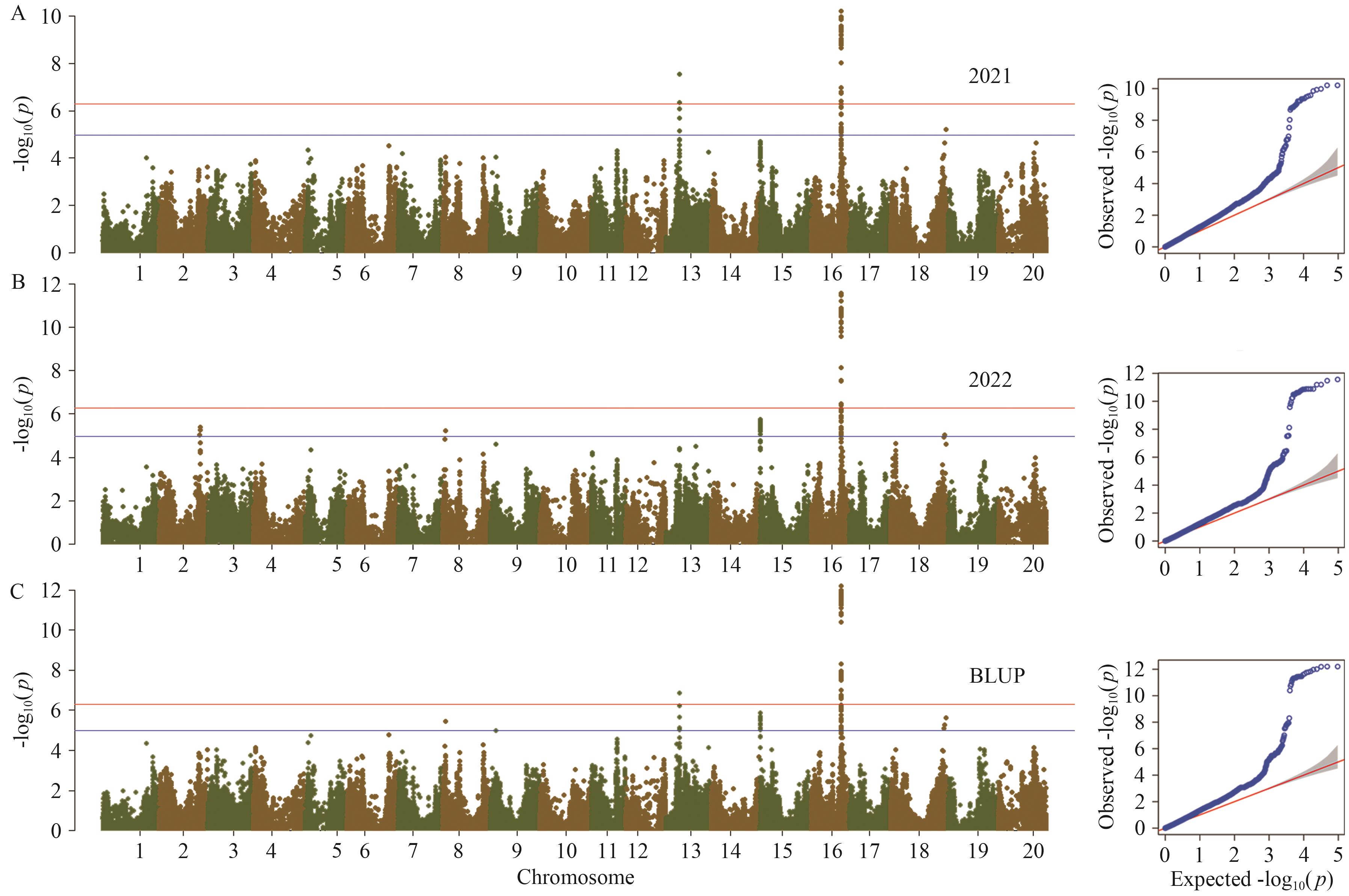
图2 裂荚性表型的GWAS分析结果和残差QQ图
Fig. 2 GWAS analysis and residual QQ plot of pod dehiscence habit
QTL名称 QTL name | SNP名称 SNP name | 染色体 Chromosome | 位置 Position/bp | 最小等位基因频率 MAF | P值 P value | 表型解释率 R2/% |
|---|---|---|---|---|---|---|
| qPdh-Chr08 | Gm08_3048312 | 8 | 3 048 312 | 0.24 | 3.50E-06 | 13.2 |
| qPdh-Chr15 | Gm15_312814 | 15 | 312 814 | 0.18 | 1.32E-06 | 10.7 |
| qPdh-Chr16(qPdh1) | Gm16_29951529 | 16 | 29 951 529 | 0.36 | 3.20E-17 | 36.6 |
| qPdh-Chr13 | Gm13_13702059 | 13 | 13 702 059 | 0.37 | 1.32E-07 | 0.5 |
| qPdh-Chr18 | Gm18_55699653 | 18 | 55 699 653 | 0.43 | 2.42E-06 | 1.8 |
表1 大豆裂荚GWAS主要关联SNP位点信息
Table 1 Main associated SNP loci in the GWAS of soybean pod dehiscence habit
QTL名称 QTL name | SNP名称 SNP name | 染色体 Chromosome | 位置 Position/bp | 最小等位基因频率 MAF | P值 P value | 表型解释率 R2/% |
|---|---|---|---|---|---|---|
| qPdh-Chr08 | Gm08_3048312 | 8 | 3 048 312 | 0.24 | 3.50E-06 | 13.2 |
| qPdh-Chr15 | Gm15_312814 | 15 | 312 814 | 0.18 | 1.32E-06 | 10.7 |
| qPdh-Chr16(qPdh1) | Gm16_29951529 | 16 | 29 951 529 | 0.36 | 3.20E-17 | 36.6 |
| qPdh-Chr13 | Gm13_13702059 | 13 | 13 702 059 | 0.37 | 1.32E-07 | 0.5 |
| qPdh-Chr18 | Gm18_55699653 | 18 | 55 699 653 | 0.43 | 2.42E-06 | 1.8 |
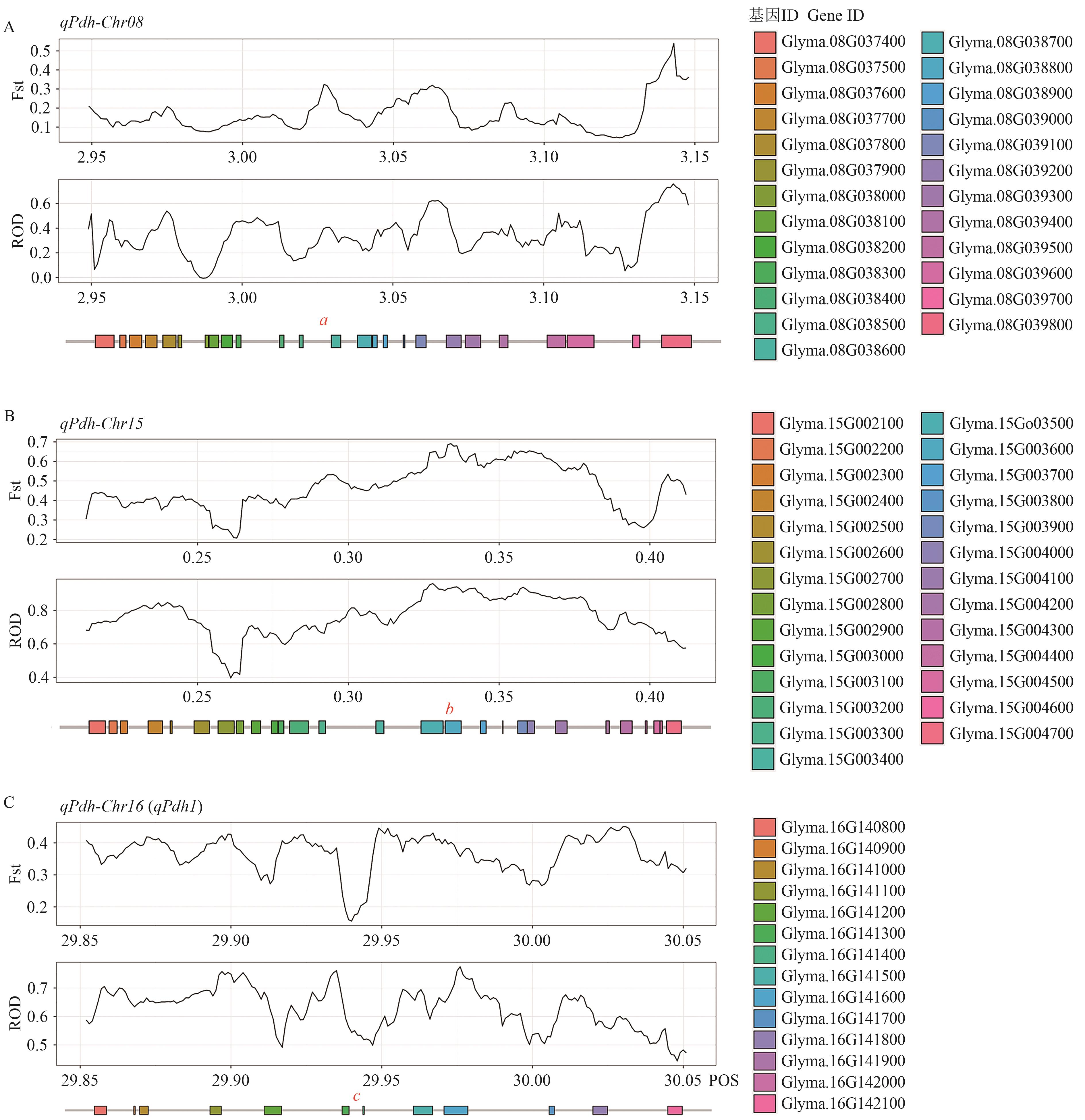
图3 栽培和野生大豆群体分化分析和相关基因
Fig. 3 Differentiation analysis of cultivated and wild soybeans population and related genes
QTL名称 QTL name | 基因 Gene | 基因注释 Gene annotation | 基因位置 Gene location |
|---|---|---|---|
| qPdh-Chr08 | Glyma.08G037400 | Tesmin/TSO1样CXC结构域,富含半胱氨酸的结构域 Tesmin/TSO1-like CXC domain, and cysteine-rich domain | Chr.8:29 50 964-2 957 163 |
| qPdh-Chr08 | Glyma.08G037500 | - | Chr.8:2 959 084-2 961 115 |
| qPdh-Chr08 | Glyma.08G037600 | - | Chr.8:2 962 310-2 966 347 |
| qPdh-Chr08 | Glyma.08G037700 | - | Chr.8:2 967 634-2 971 428 |
| qPdh-Chr08 | Glyma.08G037800 | - | Chr.8:2 973 386-2 977 716 |
| qPdh-Chr08 | Glyma.08G037900 | 动力蛋白轻链1型 Dynein light chain type 1 | Chr.8:2 978 411-2 979 627 |
| qPdh-Chr08 | Glyma.08G038000 | 无注释 No annotation | Chr.8:2 987 443-2 988 562 |
| qPdh-Chr08 | Glyma.08G038100 | S26信号肽酶,S26肽酶 S26 signal peptidase, peptidase S26 | Chr.8:2 988 728-2 991 924 |
| qPdh-Chr08 | Glyma.08G038200 | PPR重复序列 PPR repeat | Chr.8:2 992 820-2 996 514 |
| qPdh-Chr08 | Glyma.08G038300 | - | Chr.8:2 997 737-2 999 328 |
| qPdh-Chr08 | Glyma.08G038400 | 转录抑制因子 Transcriptional repressor | Chr.8:3 012 183-3 013 602 |
| qPdh-Chr08 | Glyma.08G038500 | - | Chr.8:3 018 702-3 019 975 |
| qPdh-Chr08 | Glyma.08G038600 | 黄素结合单加氧酶 Flavin-binding monooxygenase-like | Chr.8:3 029 349-3 032 467 |
| qPdh-Chr08 | Glyma.08G038700 | MVB通路中Vps4活性的调节因子 Regulator of Vps4 activity in the MVB pathway | Chr.8:3 038 024-3 042 749 |
| qPdh-Chr08 | Glyma.08G038800 | - | Chr.8:3 043 080-3 044 565 |
| qPdh-Chr08 | Glyma.08G038900 | Ras基因家族 Ras family | Chr.8:3 046 616-3 047 930 |
| qPdh-Chr08 | Glyma.08G039000 | - | Chr.8:3 053 220-3 053 733 |
| qPdh-Chr08 | Glyma.08G039100 | Frigida样蛋白 Frigida-like protein | Chr.8:3 057 438-3 060 816 |
| qPdh-Chr08 | Glyma.08G039200 | 乙醛脱氢酶家族 Aldehyde dehydrogenase family | Chr.8:3 067 517-3 072 472 |
| qPdh-Chr08 | Glyma.08G039300 | 乙醛脱氢酶家族 Aldehyde dehydrogenase family | Chr.8:3 073 843-3 078 936 |
| qPdh-Chr08 | Glyma.08G039400 | 富含亮氨酸的重复N-末端结构域 Leucine rich repeat N-terminal domain | Chr.8:3 085 136-3 088 000 |
| qPdh-Chr08 | Glyma.08G039500 | 丝氨酸激酶(Serinc) Serine incorporator (Serinc) | Chr.8:3 101 025-3 107 162 |
| qPdh-Chr08 | Glyma.08G039600 | Arf的GTP酶激活蛋白 Putative GTPase activating protein for Arf | Chr.8:3 107 698-3 116 601 |
| qPdh-Chr08 | Glyma.08G039700 | 重金属伴生域 Heavy-metal-associated domain | Chr.8:3 129 407-3 131 827 |
| qPdh-Chr08 | Glyma.08G039800 | 纤维连接蛋白Ⅲ型结构域 Fibronectin type Ⅲ domain | Chr.8:3 139 067-3 148 954 |
| qPdh-Chr15 | Glyma.15G002100 | MULE转座酶结构域 MULE transposase domain | Chr.15:213 887-219 265 |
| qPdh-Chr15 | Glyma.15G002200 | 细胞色素P450 Cytochrome P450 | Chr.15:220 497-223 162 |
| qPdh-Chr15 | Glyma.15G002300 | 真核天冬氨酸蛋白酶 Eukaryotic aspartyl protease | Chr.15:224 303-226 574 |
| qPdh-Chr15 | Glyma.15G002400 | - | Chr.15:233 459-238 218 |
| qPdh-Chr15 | Glyma.15G002500 | - | Chr.15:240 793-241 445 |
| qPdh-Chr15 | Glyma.15G002600 | 转移酶家族 Transferase family | Chr.15:248 742-253 777 |
| qPdh-Chr15 | Glyma.15G002700 | CorA样Mg2+转运蛋白 CorA-like Mg2+ transporter protein | Chr.15:256 675-262 022 |
| qPdh-Chr15 | Glyma.15G002800 | PPR重复 PPR repeat | Chr.15:262 741-265 142 |
| qPdh-Chr15 | Glyma.15G002900 | 蛋白激酶结构域 Protein kinase domain | Chr.15:267 776-270 825 |
| qPdh-Chr15 | Glyma.15G003000 | - | Chr.15:274 385-276 541 |
| qPdh-Chr15 | Glyma.15G003100 | 3-氧乙酰基-[酰基载体蛋白(ACP)]合酶Ⅲ 4-3-Oxoacyl-[acyl-carrier-protein (ACP)] synthase Ⅲ | Chr.15:276 702-278 479 |
| qPdh-Chr15 | Glyma.15G003200 | - | Chr.15:280 413-286 646 |
| qPdh-Chr15 | Glyma.15G003300 | WRKY DNA结合结构域 WRKY DNA-binding domain | Chr.15:290 145-292 323 |
| qPdh-Chr15 | Glyma.15G003400 | NAF结构域 NAF domain | Chr.15:309 074-311 683 |
| qPdh-Chr15 | Glyma.15G003500 | 天然抗性相关巨噬细胞蛋白 Natural resistance-associated macrophage protein | Chr.15:324 025-331 370 |
| qPdh-Chr15 | Glyma.15G003600 | NAD依赖性差向异构酶/脱水酶家族 DAD dependent epimerase/dehydratase family | Chr.15:332 065-337 381 |
| qPdh-Chr15 | Glyma.15G003700 | 核糖体蛋白L7Ae/L30e/S12e/Gadd45家族 Ribosomal protein L7Ae/L30e/S12e/Gadd45 family | Chr.15:343 771-345 651 |
| qPdh-Chr15 | Glyma.15G003800 | 光系统I反应中心亚基Ⅷ Photosystem I reaction centre subunit Ⅷ | Chr.15:351 123-351 227 |
| qPdh-Chr15 | Glyma.15G003900 | 主要内在蛋白质 Major intrinsic protein | Chr.15:356 131-360 422 |
| qPdh-Chr15 | Glyma.15G004000 | - | Chr.15:359 275-361 686 |
| qPdh-Chr15 | Glyma.15G004100 | - | Chr.15:368 686-372 503 |
| qPdh-Chr15 | Glyma.15G004200 | AP2结构域 AP2 domain | Chr.15:385 421-386 513 |
| qPdh-Chr15 | Glyma.15G004300 | 卤代酸脱卤酶样水解酶;阳离子转运蛋白/ATP酶;N端 Haloacid dehalogenase-like hydrolase; cation transporter/ATPase; N-terminus | Chr.15:390 297-394 072 |
| qPdh-Chr15 | Glyma.15G004400 | - | Chr.15:398 466-399 046 |
| qPdh-Chr15 | Glyma.15G004500 | 双链RNA结合基序 Double-stranded RNA binding motif | Chr.15:401 329-403 329 |
| qPdh-Chr15 | Glyma.15G004600 | - | Chr.15:403 355-404 122 |
| qPdh-Chr15 | Glyma.15G004700 | 蛋白酪氨酸激酶 Protein tyrosine kinase | Chr.15:405 502-410 429 |
| qPdh-Chr16(qPdh1) | Glyma.16G141400 | - | Chr.16:29 943 929-29 944 387 |
表2 大豆裂荚候选基因信息
Table 2 Candidate genes information of pod dehiscence habit in soybean
QTL名称 QTL name | 基因 Gene | 基因注释 Gene annotation | 基因位置 Gene location |
|---|---|---|---|
| qPdh-Chr08 | Glyma.08G037400 | Tesmin/TSO1样CXC结构域,富含半胱氨酸的结构域 Tesmin/TSO1-like CXC domain, and cysteine-rich domain | Chr.8:29 50 964-2 957 163 |
| qPdh-Chr08 | Glyma.08G037500 | - | Chr.8:2 959 084-2 961 115 |
| qPdh-Chr08 | Glyma.08G037600 | - | Chr.8:2 962 310-2 966 347 |
| qPdh-Chr08 | Glyma.08G037700 | - | Chr.8:2 967 634-2 971 428 |
| qPdh-Chr08 | Glyma.08G037800 | - | Chr.8:2 973 386-2 977 716 |
| qPdh-Chr08 | Glyma.08G037900 | 动力蛋白轻链1型 Dynein light chain type 1 | Chr.8:2 978 411-2 979 627 |
| qPdh-Chr08 | Glyma.08G038000 | 无注释 No annotation | Chr.8:2 987 443-2 988 562 |
| qPdh-Chr08 | Glyma.08G038100 | S26信号肽酶,S26肽酶 S26 signal peptidase, peptidase S26 | Chr.8:2 988 728-2 991 924 |
| qPdh-Chr08 | Glyma.08G038200 | PPR重复序列 PPR repeat | Chr.8:2 992 820-2 996 514 |
| qPdh-Chr08 | Glyma.08G038300 | - | Chr.8:2 997 737-2 999 328 |
| qPdh-Chr08 | Glyma.08G038400 | 转录抑制因子 Transcriptional repressor | Chr.8:3 012 183-3 013 602 |
| qPdh-Chr08 | Glyma.08G038500 | - | Chr.8:3 018 702-3 019 975 |
| qPdh-Chr08 | Glyma.08G038600 | 黄素结合单加氧酶 Flavin-binding monooxygenase-like | Chr.8:3 029 349-3 032 467 |
| qPdh-Chr08 | Glyma.08G038700 | MVB通路中Vps4活性的调节因子 Regulator of Vps4 activity in the MVB pathway | Chr.8:3 038 024-3 042 749 |
| qPdh-Chr08 | Glyma.08G038800 | - | Chr.8:3 043 080-3 044 565 |
| qPdh-Chr08 | Glyma.08G038900 | Ras基因家族 Ras family | Chr.8:3 046 616-3 047 930 |
| qPdh-Chr08 | Glyma.08G039000 | - | Chr.8:3 053 220-3 053 733 |
| qPdh-Chr08 | Glyma.08G039100 | Frigida样蛋白 Frigida-like protein | Chr.8:3 057 438-3 060 816 |
| qPdh-Chr08 | Glyma.08G039200 | 乙醛脱氢酶家族 Aldehyde dehydrogenase family | Chr.8:3 067 517-3 072 472 |
| qPdh-Chr08 | Glyma.08G039300 | 乙醛脱氢酶家族 Aldehyde dehydrogenase family | Chr.8:3 073 843-3 078 936 |
| qPdh-Chr08 | Glyma.08G039400 | 富含亮氨酸的重复N-末端结构域 Leucine rich repeat N-terminal domain | Chr.8:3 085 136-3 088 000 |
| qPdh-Chr08 | Glyma.08G039500 | 丝氨酸激酶(Serinc) Serine incorporator (Serinc) | Chr.8:3 101 025-3 107 162 |
| qPdh-Chr08 | Glyma.08G039600 | Arf的GTP酶激活蛋白 Putative GTPase activating protein for Arf | Chr.8:3 107 698-3 116 601 |
| qPdh-Chr08 | Glyma.08G039700 | 重金属伴生域 Heavy-metal-associated domain | Chr.8:3 129 407-3 131 827 |
| qPdh-Chr08 | Glyma.08G039800 | 纤维连接蛋白Ⅲ型结构域 Fibronectin type Ⅲ domain | Chr.8:3 139 067-3 148 954 |
| qPdh-Chr15 | Glyma.15G002100 | MULE转座酶结构域 MULE transposase domain | Chr.15:213 887-219 265 |
| qPdh-Chr15 | Glyma.15G002200 | 细胞色素P450 Cytochrome P450 | Chr.15:220 497-223 162 |
| qPdh-Chr15 | Glyma.15G002300 | 真核天冬氨酸蛋白酶 Eukaryotic aspartyl protease | Chr.15:224 303-226 574 |
| qPdh-Chr15 | Glyma.15G002400 | - | Chr.15:233 459-238 218 |
| qPdh-Chr15 | Glyma.15G002500 | - | Chr.15:240 793-241 445 |
| qPdh-Chr15 | Glyma.15G002600 | 转移酶家族 Transferase family | Chr.15:248 742-253 777 |
| qPdh-Chr15 | Glyma.15G002700 | CorA样Mg2+转运蛋白 CorA-like Mg2+ transporter protein | Chr.15:256 675-262 022 |
| qPdh-Chr15 | Glyma.15G002800 | PPR重复 PPR repeat | Chr.15:262 741-265 142 |
| qPdh-Chr15 | Glyma.15G002900 | 蛋白激酶结构域 Protein kinase domain | Chr.15:267 776-270 825 |
| qPdh-Chr15 | Glyma.15G003000 | - | Chr.15:274 385-276 541 |
| qPdh-Chr15 | Glyma.15G003100 | 3-氧乙酰基-[酰基载体蛋白(ACP)]合酶Ⅲ 4-3-Oxoacyl-[acyl-carrier-protein (ACP)] synthase Ⅲ | Chr.15:276 702-278 479 |
| qPdh-Chr15 | Glyma.15G003200 | - | Chr.15:280 413-286 646 |
| qPdh-Chr15 | Glyma.15G003300 | WRKY DNA结合结构域 WRKY DNA-binding domain | Chr.15:290 145-292 323 |
| qPdh-Chr15 | Glyma.15G003400 | NAF结构域 NAF domain | Chr.15:309 074-311 683 |
| qPdh-Chr15 | Glyma.15G003500 | 天然抗性相关巨噬细胞蛋白 Natural resistance-associated macrophage protein | Chr.15:324 025-331 370 |
| qPdh-Chr15 | Glyma.15G003600 | NAD依赖性差向异构酶/脱水酶家族 DAD dependent epimerase/dehydratase family | Chr.15:332 065-337 381 |
| qPdh-Chr15 | Glyma.15G003700 | 核糖体蛋白L7Ae/L30e/S12e/Gadd45家族 Ribosomal protein L7Ae/L30e/S12e/Gadd45 family | Chr.15:343 771-345 651 |
| qPdh-Chr15 | Glyma.15G003800 | 光系统I反应中心亚基Ⅷ Photosystem I reaction centre subunit Ⅷ | Chr.15:351 123-351 227 |
| qPdh-Chr15 | Glyma.15G003900 | 主要内在蛋白质 Major intrinsic protein | Chr.15:356 131-360 422 |
| qPdh-Chr15 | Glyma.15G004000 | - | Chr.15:359 275-361 686 |
| qPdh-Chr15 | Glyma.15G004100 | - | Chr.15:368 686-372 503 |
| qPdh-Chr15 | Glyma.15G004200 | AP2结构域 AP2 domain | Chr.15:385 421-386 513 |
| qPdh-Chr15 | Glyma.15G004300 | 卤代酸脱卤酶样水解酶;阳离子转运蛋白/ATP酶;N端 Haloacid dehalogenase-like hydrolase; cation transporter/ATPase; N-terminus | Chr.15:390 297-394 072 |
| qPdh-Chr15 | Glyma.15G004400 | - | Chr.15:398 466-399 046 |
| qPdh-Chr15 | Glyma.15G004500 | 双链RNA结合基序 Double-stranded RNA binding motif | Chr.15:401 329-403 329 |
| qPdh-Chr15 | Glyma.15G004600 | - | Chr.15:403 355-404 122 |
| qPdh-Chr15 | Glyma.15G004700 | 蛋白酪氨酸激酶 Protein tyrosine kinase | Chr.15:405 502-410 429 |
| qPdh-Chr16(qPdh1) | Glyma.16G141400 | - | Chr.16:29 943 929-29 944 387 |
| 1 | Funatsuki H, Hajika M, Yamada T, et al. Mapping and use of QTLs controlling pod dehiscence in soybean[J]. Breed Sci, 2012, 61(5): 554-558. |
| 2 | Ogutcen E, Pandey A, Khan MK, et al. Pod shattering: a homologous series of variation underlying domestication and an avenue for crop improvement[J]. Agronomy, 2018, 8(8): 137. |
| 3 | Di Vittori V, Gioia T, Rodriguez M, et al. Convergent evolution of the seed shattering trait[J]. Genes, 2019, 10(1): 68. |
| 4 | Tsuchiya T. Studies on shattering resistance in soybean breeding[J]. Report of Hokkaido Prefectural Agricultural Experiment Station, 1986, 58: 1-53. |
| 5 | Bailey MA, Mian MAR, Carter TE, et al. Pod dehiscence of soybean: Identification of quantitative trait loci[J]. J Hered, 1997, 88(2): 152-154. |
| 6 | Funatsuki H, Ishimoto M, Tsuji H, et al. Simple sequence repeat markers linked to a major QTL controlling pod shattering in soybean [J]. Plant Breed, 2006, 125(2): 195-197. |
| 7 | Liu BH, Fujita T, Yan ZH, et al. QTL mapping of domestication-related traits in soybean (Glycine max)[J]. Ann Bot, 2007, 100(5): 1027-1038. |
| 8 | Suzuki M, Fujino K, Nakamoto Y, et al. Fine mapping and development of DNA markers for the qPDH1 locus associated with pod dehiscence in soybean[J]. Mol Breed, 2010, 25(3): 407-418. |
| 9 | Lee JS, Kim KR, Ha BK, et al. Identification of SNPs tightly linked to the QTL for pod shattering in soybean[J]. Mol Breed, 2017, 37(4): 54. |
| 10 | Miranda C, Culp C, Škrabišová M, et al. Molecular tools for detecting Pdh1 can improve soybean breeding efficiency by reducing yield losses due to pod shatter[J]. Mol Breed, 2019, 39(2): 27. |
| 11 | Funatsuki H, Suzuki M, Hirose A, et al. Molecular basis of a shattering resistance boosting global dissemination of soybean[J]. Proc Natl Acad Sci USA, 2014, 111(50): 17797-17802 |
| 12 | Yong B, Zhu WW, Wei SM, et al. Parallel selection of loss-of-function alleles of Pdh1 orthologous genes in warm-season legumes for pod indehiscence and plasticity is related to precipitation[J]. New Phytol, 2023, 240(2): 863-879. |
| 13 | Ma XF, Xu WY, Liu T, et al. Functional characterization of soybean (Glycine max) DIRIGENT genes reveals an important role of GmDIR27 in the regulation of pod dehiscence[J]. Genomics, 2021, 113(1): 979-990. |
| 14 | Zhang JP, Singh AK. Genetic control and geo-climate adaptation of pod dehiscence provide novel insights into soybean domestication[J]. G3, 2020, 10(2): 545-554. |
| 15 | Dong Y, Yang X, Liu J, et al. Pod shattering resistance associated with domestication is mediated by a NAC gene in soybean[J]. Nat Commun, 2014, 5: 3352. |
| 16 | 韩德志, 任玉龙, 郭勇, 等. 大豆炸荚发生规律及分子遗传基础[J]. 遗传, 2015, 37(6): 535-543. |
| Han DZ, Ren YL, Guo Y, et al. Occurrence characteristics and molecular genetic basis of pod shattering in soybean[J]. Heredity, 2015, 37(6): 535-543. | |
| 17 | Fuller DQ. Contrasting patterns in crop domestication and domestication rates: Recent archaeobotanical insights from the old world[J]. Ann Bot, 2007, 100(5): 903-924. |
| 18 | 张跃进, 马赛斐, 高启云, 等. 黄淮流域主栽大豆品种炸荚性研究[J]. 河南农业科学, 2006, 35(6): 56-59. |
| Zhang YJ, Ma SF, Gao QY, et al. Study on the pod shattering of main soybean varieties of Huanghuai area[J] J Henan Agric Sci, 2006, 35(6): 56-59. | |
| 19 | 窦玲, 郝青南, 杨中路, 等. 大豆抗裂荚基因pdh1在长江中下游区域联合试验品种中的分布[J]. 中国油料作物学报, 2023, 45(4): 704-710. |
| Dou L, Hao QN, Yang ZL, et al. Distribution of pod shattering resistant gene pdh1 in varieties for multiple variety tests in Yangtze River regions[J]. Chin J Oil Crop Sci, 2023, 45(4): 704-710. | |
| 20 | Knapp SJ, Stroup WW, Ross WM. Exact confidence intervals for heritability on a progeny mean basis[J]. Crop Sci, 1985, 25(1): 192-194. |
| 21 | Wang JB, Zhang ZW. GAPIT version 3: Boosting power and accuracy for genomic association and prediction[J]. Genomics Proteomics Bioinformatics, 2021, 19(4): 629-640. |
| 22 | Wang M, Yan JB, Zhao JR, et al. Genome-wide association study (GWAS) of resistance to head smut in maize[J]. Plant Sci, 2012, 196: 125-131. |
| 23 | Zhang HY, Jiang H, Hu ZB, et al. Development of a versatile resource for post-genomic research through consolidating and characterizing 1500 diverse wild and cultivated soybean genomes[J]. BMC Genomics, 2022, 23(1): 250. |
| 24 | Jia J, Wang H, Cai ZD, et al. Identification and validation of stable and novel quantitative trait loci for pod shattering in soybean [Glycine max (L.) Merr.][J]. J Intege Agric, 2022, 21(11): 3169-3184. |
| 25 | Hu DZ, Kan GZ, Hu W, et al. Identification of loci and candidate genes responsible for pod dehiscence in soybean via genome-wide association analysis across multiple environments[J]. Front Plant Sci, 2019, 21(10): 811. |
| 26 | 余泓, 李家洋. 是金子无论在何处都发光: 玉米和水稻驯化中的趋同选择[J]. 植物学报, 2022, 57(2): 153-156. |
| Yu H, Li JY. The gold will glitter wherever it is: Convergent selection in domestication of maize and rice[J]. Chin Bull Bot, 2022, 57(2): 153-156. | |
| 27 | Zhao Y, Christensen SK, Fankhauser C, et al. A role for flavin monooxygenase-like enzymes in auxin biosynthesis[J]. Science, 2001, 291(5502): 306-309. |
| 28 | Mir Derikvand M, Sierra JB, Ruel K, et al. Redirection of the phenylpropanoid pathway to feruloyl malate in Arabidopsis mutants deficient for cinnamoyl-CoA reductase 1[J]. Planta, 2008, 227(5): 943-956. |
| [1] | 于静, 于桂爽, 孙昊杰, 姜春姣, 苑广迪, 杨珍, 王志伟, 王超, 王传堂. 花生种用品质影响因素及相关标记研究[J]. 生物技术通报, 2025, 41(2): 284-294. |
| [2] | 贺涵, 刘传和, 喻梦凡, 袁梦萍, 魏岳荣, 杨敏, 邝瑞彬, 周陈平, 吴夏明, 徐泽. 基于重测序的菠萝基因组InDel标记的开发[J]. 生物技术通报, 2025, 41(2): 65-76. |
| [3] | 刘克寒, 杨升辉, 黄巧云, 崔文靖. 黑龙江大豆根瘤菌及根际促共生菌株的筛选及应用[J]. 生物技术通报, 2025, 41(1): 252-262. |
| [4] | 毛向红, 卢瑶, 范向斌, 杜培兵, 白小东. 基于SSR荧光标记毛细管电泳的马铃薯品种遗传多样性分析及分子身份证构建[J]. 生物技术通报, 2024, 40(9): 131-140. |
| [5] | 孙志勇, 杜怀东, 刘阳, 马嘉欣, 于雪然, 马伟, 姚鑫杰, 王敏, 李培富. 水稻籽粒γ-氨基丁酸含量的全基因组关联分析[J]. 生物技术通报, 2024, 40(8): 53-62. |
| [6] | 武帅, 辛燕妮, 买春海, 穆晓娅, 王敏, 岳爱琴, 赵晋忠, 吴慎杰, 杜维俊, 王利祥. 大豆GS基因家族全基因组鉴定及胁迫响应分析[J]. 生物技术通报, 2024, 40(8): 63-73. |
| [7] | 高萌萌, 赵天宇, 焦馨悦, 林春晶, 关哲允, 丁孝羊, 孙妍妍, 张春宝. 大豆细胞质雄性不育系及其恢复系的比较转录组分析[J]. 生物技术通报, 2024, 40(7): 137-149. |
| [8] | 王芳, 于璐, 齐泽铮, 周长军, 于吉东. 大豆镰刀菌根腐病拮抗菌的筛选及生防效果[J]. 生物技术通报, 2024, 40(7): 216-225. |
| [9] | 白志元, 徐菲, 杨午, 王明贵, 杨玉花, 张海平, 张瑞军. 大豆细胞质雄性不育弱恢复型杂种F1育性转变的转录组分析[J]. 生物技术通报, 2024, 40(6): 134-142. |
| [10] | 孔小平, 陈利文, 刘思思, 严湘萍. 胡萝卜抽薹相关性状全基因组关联分析[J]. 生物技术通报, 2024, 40(5): 120-130. |
| [11] | 李思琪, 张文臣, 杨柳, 付庆新, 洪新, 张海旺. 基于SSR标记的文冠果遗传多样性分析及指纹图谱构建[J]. 生物技术通报, 2024, 40(5): 74-83. |
| [12] | 娄银, 高浩竣, 王茜, 牛景萍, 王敏, 杜维俊, 岳爱琴. 大豆GmHMGS基因的鉴定及表达模式分析[J]. 生物技术通报, 2024, 40(4): 110-121. |
| [13] | 李晴, 石雨荷, 朱珏, 李晓玲, 侯超文, 童巧珍. 基于SCoT分子标记分析白术种质资源遗传多样性及DNA指纹图谱构建[J]. 生物技术通报, 2024, 40(11): 142-151. |
| [14] | 刘奕君, 闫伟, 何禹璇, 董立明, 龙丽坤, 李飞武. 转基因大豆多靶标质粒DNA标准分子的研制及应用[J]. 生物技术通报, 2024, 40(11): 169-183. |
| [15] | 李炯珊, 杨泽, 闫星, 刘义珍, 郭宇双, 薛金爱, 孙希平, 季春丽, 张春辉, 李润植. 四尾栅藻提高大豆草甘膦抗性及促生效应分析[J]. 生物技术通报, 2024, 40(11): 236-247. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||