







Biotechnology Bulletin ›› 2024, Vol. 40 ›› Issue (9): 270-281.doi: 10.13560/j.cnki.biotech.bull.1985.2024-0050
Previous Articles Next Articles
LIU Ding-rui1,2( ), GUO Hong-guang1,2(), GONG Kai-yi1,2
), GUO Hong-guang1,2(), GONG Kai-yi1,2
Received:2024-01-15
Online:2024-09-26
Published:2024-10-12
Contact:
GUO Hong-guang
E-mail:1786013906@qq.com;guohg_tyut@163.com
LIU Ding-rui, GUO Hong-guang, GONG Kai-yi. Metagenomic and Metatranscriptomic Analysis of Methanogenesis from Coal Degradation by Compounded Microflora[J]. Biotechnology Bulletin, 2024, 40(9): 270-281.

Fig. 1 Biomethane yield in CM and RI
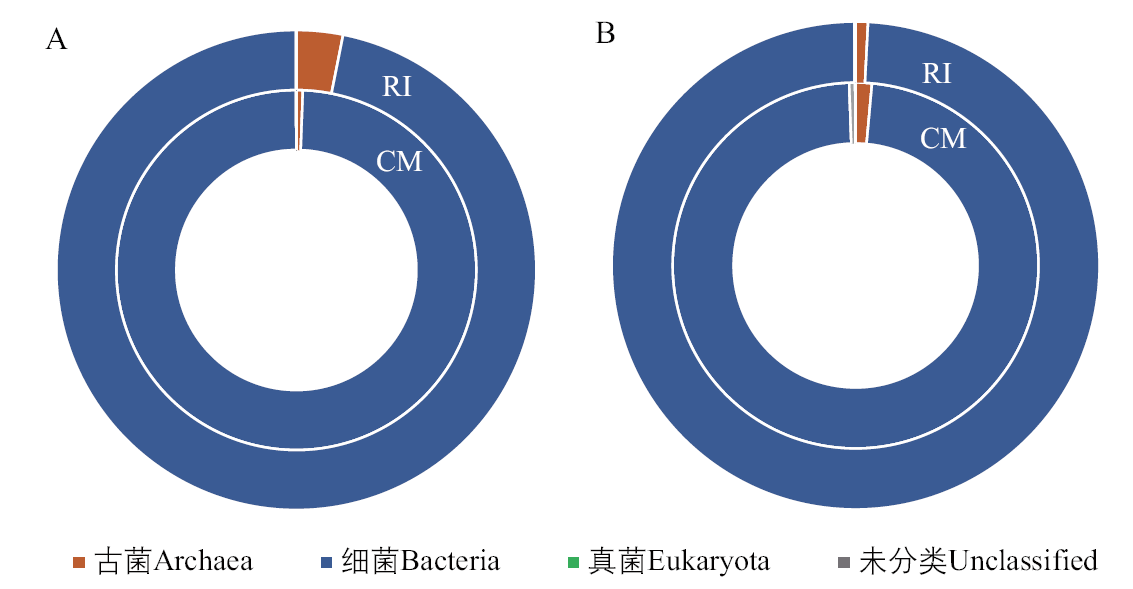
Fig. 2 Distribution of RI and CM at domain level A: Distribution of RI and CM at the level of metagenomic domains; B: Distribution of RI and CM at the level of metatranscriptomic domains
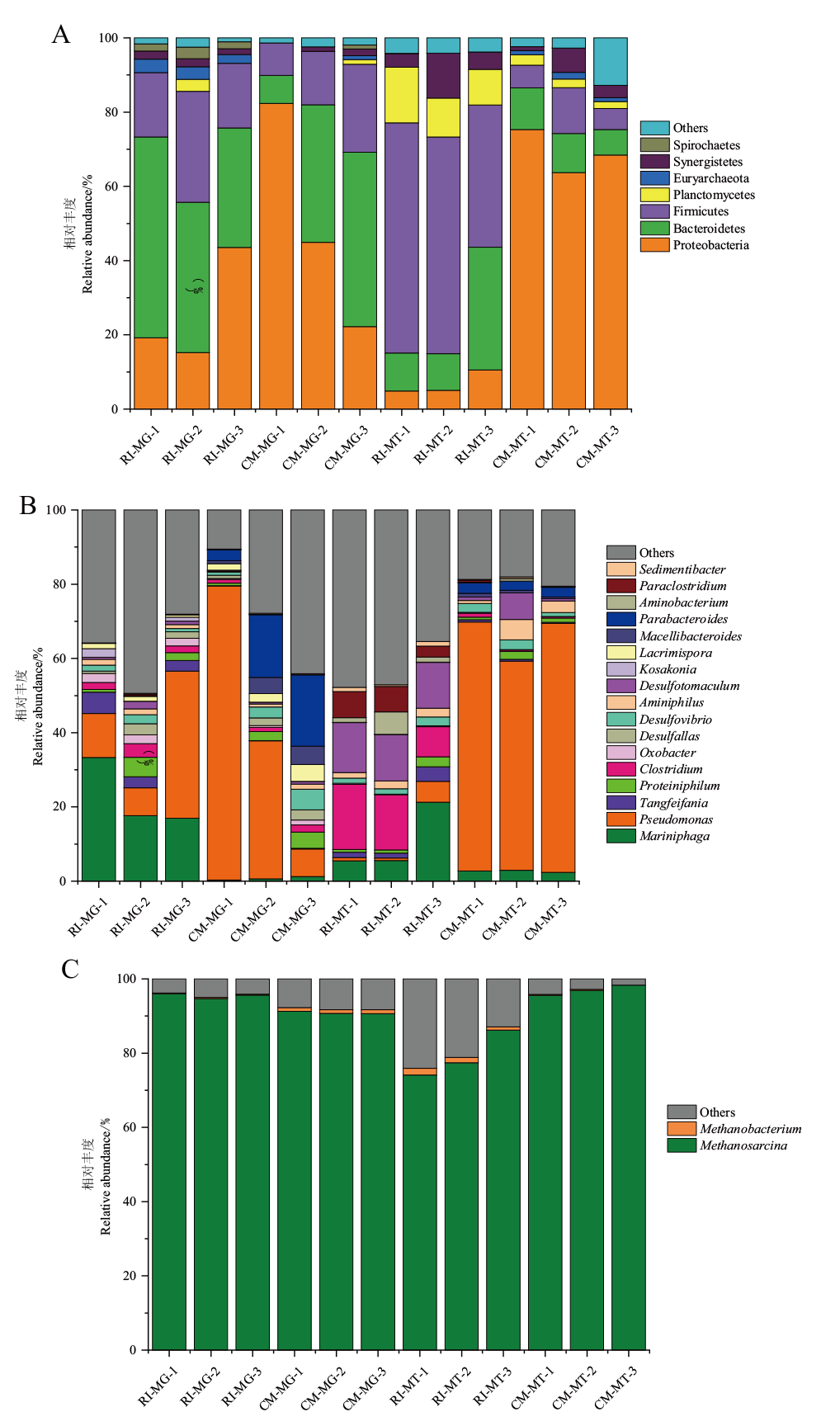
Fig. 3 Microbial composition maps of three parallel groups of RI and CM samples at phylum(A), bacterial genus(B), and archaeal genus(C) MG: Metagenomic; MT: metatranscriptomic,the same below
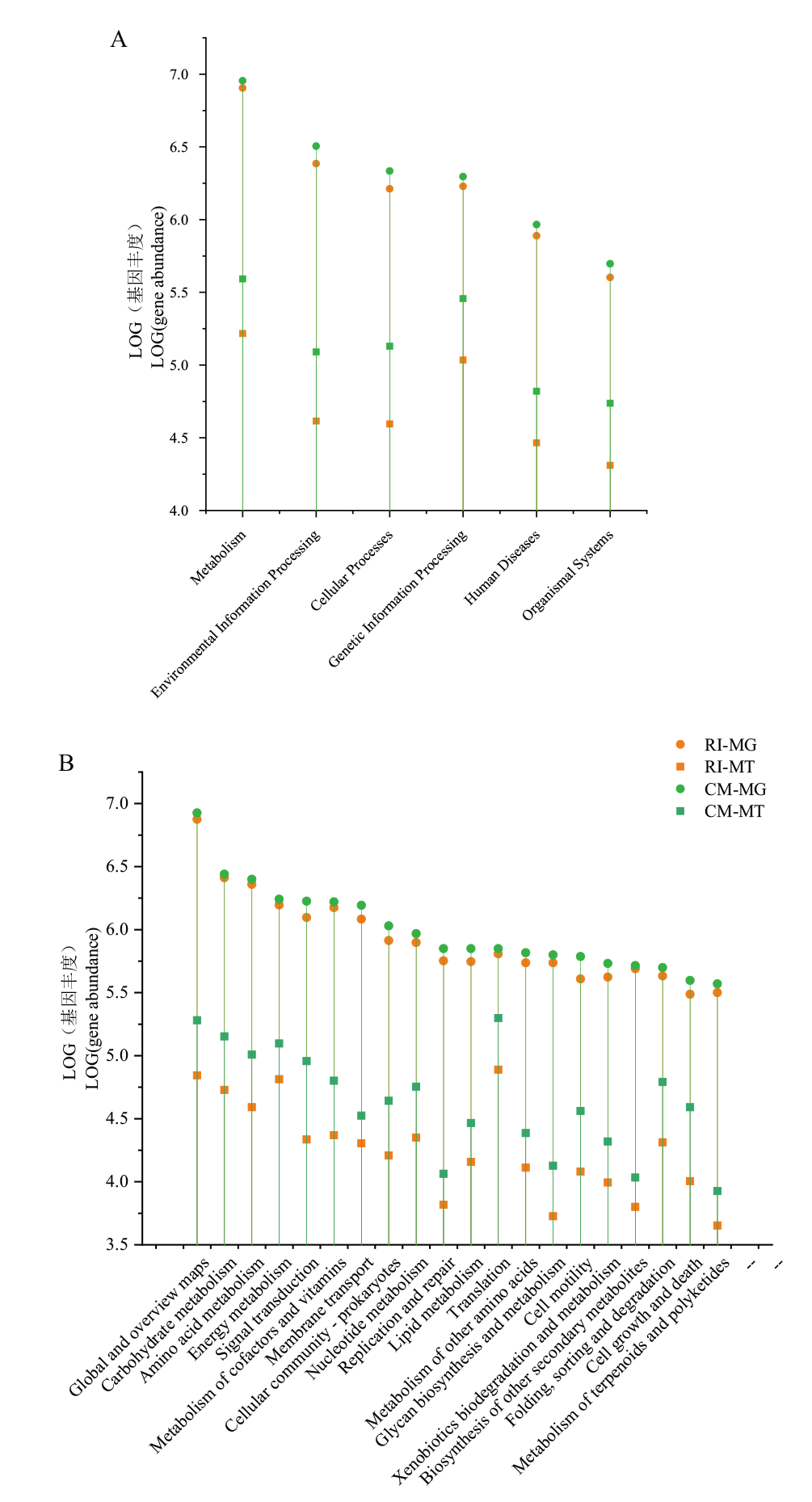
Fig. 4 Absolute abundance of gene presence and expression between primary(A)and secondary(B)metabolic systems in bioreactor RI and CM The Y-axis scale refers to the logarithm of sequence abundance(sequences with annotations)for each category
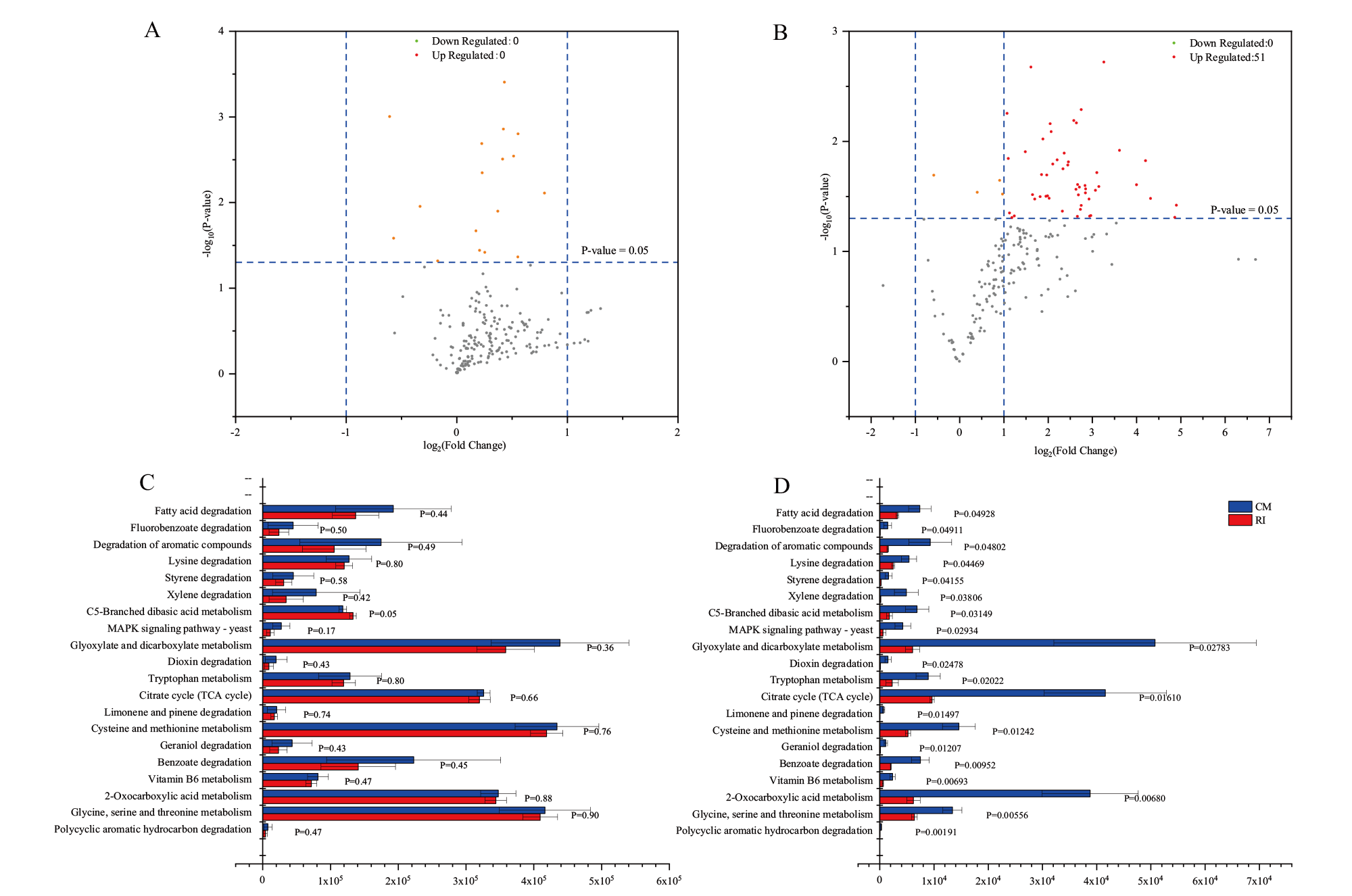
Fig. 5 Differential analysis of RI and CM metabolic pathways on tertiary pathways A:Volcano plot of KEGG in metagenomic in tertiary pathways. B: Volcano plot of KEGG in metatranscriptomic in tertiary pathways. C: Comparison of abundance of coal-degrading methane-producing metabolic pathways in metagenomic. D: Comparison of abundance of coal-degrading methane-producing metabolic pathways in metatranscriptomic

Fig. 6 Aromatic compound metabolic pathways and plots of gene abundance and gene expression abundance The bar graphs refer to the logarithm of abundance and expression abundance for each gene, the same below

Fig. 7 Pyruvate metabolic pathways and plots of gene abundance and gene expression abundance
| 模块Module | RI-MG | RI-MT | CM-MG | CM-MT | 功能描述 Function |
|---|---|---|---|---|---|
| M00422 | 7578 | 1453.027 | 2252 | 6649.955 | Acetyl-CoA pathway, CO2 => acetyl-CoA |
| M00569 | 15455.33 | 114.273 | 34242 | 2434.298 | Catechol meta-cleavage, catechol => acetyl-CoA / 4-methylcatechol => propanoyl-CoA |
| M00036 | 79514 | 704.217 | 98381.33 | 1709.537 | Leucine degradation, leucine => acetoacetate + acetyl-CoA |
| M00013 | 10171.33 | 16.876 | 21789.33 | 957.577 | Malonate semialdehyde pathway, propanoyl-CoA => acetyl-CoA |
| M00032 | 32807.33 | 343.8167 | 37454.67 | 2953.905 | Lysine degradation, lysine => saccharopine => acetoacetyl-CoA |
| M00307 | 70726 | 1861.692 | 64192 | 3460.332 | Pyruvate oxidation, pyruvate => acetyl-CoA |
| M00086 | 42996 | 780.0583 | 50578 | 1389.26 | Beta-oxidation, acyl-CoA synthesis |
Table 1 Main production pathways of acetyl coenzyme A and abundance of genes and expression abundance of genes in the routes
| 模块Module | RI-MG | RI-MT | CM-MG | CM-MT | 功能描述 Function |
|---|---|---|---|---|---|
| M00422 | 7578 | 1453.027 | 2252 | 6649.955 | Acetyl-CoA pathway, CO2 => acetyl-CoA |
| M00569 | 15455.33 | 114.273 | 34242 | 2434.298 | Catechol meta-cleavage, catechol => acetyl-CoA / 4-methylcatechol => propanoyl-CoA |
| M00036 | 79514 | 704.217 | 98381.33 | 1709.537 | Leucine degradation, leucine => acetoacetate + acetyl-CoA |
| M00013 | 10171.33 | 16.876 | 21789.33 | 957.577 | Malonate semialdehyde pathway, propanoyl-CoA => acetyl-CoA |
| M00032 | 32807.33 | 343.8167 | 37454.67 | 2953.905 | Lysine degradation, lysine => saccharopine => acetoacetyl-CoA |
| M00307 | 70726 | 1861.692 | 64192 | 3460.332 | Pyruvate oxidation, pyruvate => acetyl-CoA |
| M00086 | 42996 | 780.0583 | 50578 | 1389.26 | Beta-oxidation, acyl-CoA synthesis |
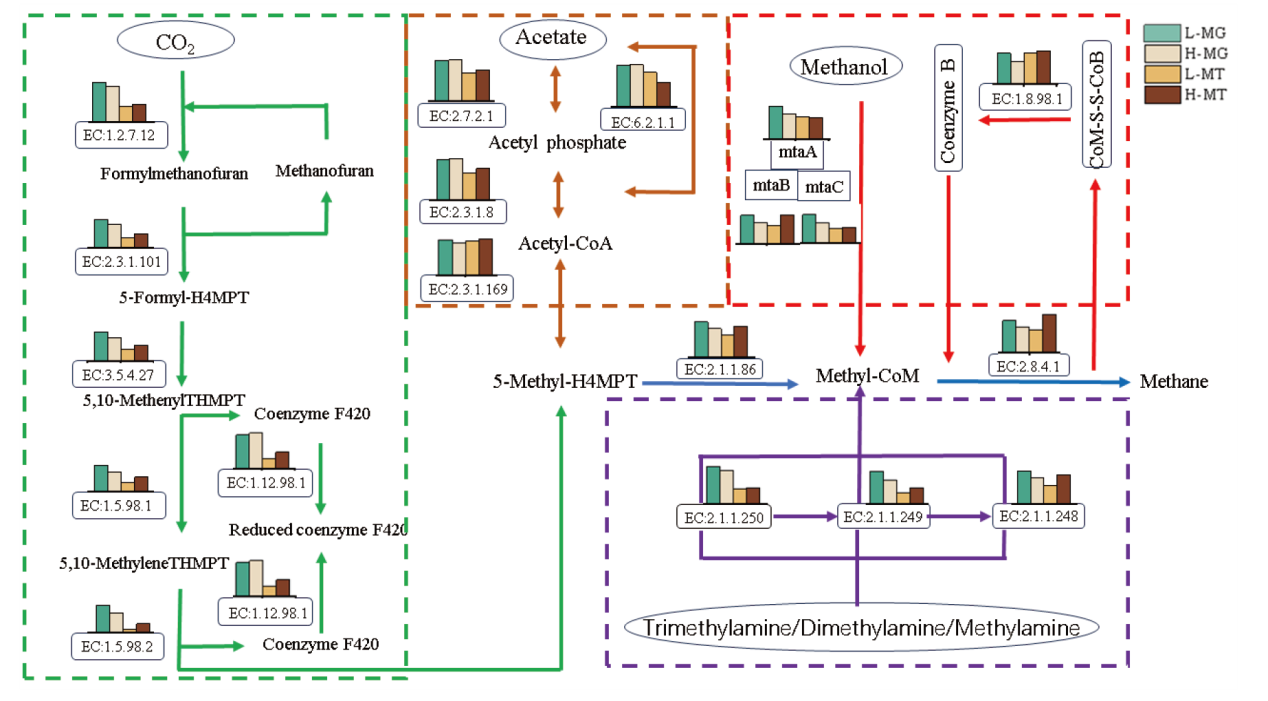
Fig. 8 Methane metabolic pathways and plots of gene abundance and gene expression abundance
| 样本名称Sample | M00357 | M00356 | M00567 | M00563 |
|---|---|---|---|---|
| RI-MG | 72573 | 48854 | 50116 | 46484 |
| RI-MT | 7123 | 1821 | 2040 | 1703 |
| CM-MG | 63086 | 28250 | 30324 | 28090 |
| CM-MT | 18937 | 12936 | 11077 | 10457 |
Table 2 Gene content and expression of each pathway of methane production
| 样本名称Sample | M00357 | M00356 | M00567 | M00563 |
|---|---|---|---|---|
| RI-MG | 72573 | 48854 | 50116 | 46484 |
| RI-MT | 7123 | 1821 | 2040 | 1703 |
| CM-MG | 63086 | 28250 | 30324 | 28090 |
| CM-MT | 18937 | 12936 | 11077 | 10457 |
| [1] |
徐凤银, 侯伟, 熊先钺, 等. 中国煤层气产业现状与发展战略[J]. 石油勘探与开发, 2023, 50(4): 669-682.
doi: 10.11698/PED.20220856 |
| Xu FY, Hou W, Xiong XY, et al. The status and development strategy of coalbed methane industry in China[J]. Petrol Explor Dev, 2023, 50(4): 669-682. | |
| [2] | 范鹏鹏, 刘晓, 陈贞龙, 等. 中美煤层气勘探开发现状对比及启示[J]. 现代化工, 2023, 43(8): 22-25, 30. |
| Fan PP, Liu X, Chen ZL, et al. Comparison and enlightenment of current situation of coalbed methane exploration and development between China and the United States[J]. Mod Chem Ind, 2023, 43(8): 22-25, 30. | |
| [3] | 薛建英. 加强企业用地保障优化矿业权登记管理山西加大煤层气勘查开采支持力度[J]. 华北自然资源, 2023(5): 161. |
| Xue JY. Strengthen enterprise land security, optimize mining right registration management, and increase support for coalbed methane exploration and exploitation in Shanxi[J]. Huabei Nat Resour, 2023(5): 161. | |
| [4] | 苏现波, 夏大平, 赵伟仲, 等. 煤层气生物工程研究进展[J]. 煤炭科学技术, 2020, 48(6): 1-30. |
| Su XB, Xia DP, Zhao WZ, et al. Research advances of coalbed gas bioengineering[J]. Coal Sci Technol, 2020, 48(6): 1-30. | |
| [5] | Zhang Y, Zhang H, Li Y, et al. Study on increasing production of bio-methane from middle/high rank coal pretreated with hydrogen peroxide[J]. Coal Science and Technology, 2019, 47(09): 262-7. |
| [6] | Zhang J, Zhang H, Wang K, et al. Research progress of pretreatment technology for coal biotransformation[J]. Coal Technology, 2016, 35(11): 317-9. |
| [7] | Gong KY, Zhang YX, Guo HG, et al. Enhancing biomethane production from lignite by an anaerobic polycyclic aromatic hydrocarbon degrading fungal flora enriched from produced water[J]. Front Microbiol, 2022, 13: 899863. |
| [8] | Guo HG, Chen C, Liang WG, et al. Enhanced biomethane production from anthracite by application of an electric field[J]. Int J Coal Geol, 2020, 219: 103393. |
| [9] | Guo HG, Cheng YT, Huang ZX, et al. Factors affecting co-degradation of coal and straw to enhance biogenic coalbed methane[J]. Fuel, 2019, 244: 240-246. |
| [10] | Liu XS, Liu YJ, Tang H, et al. Microbial metabolism regulation on the efficient degradation of aromatic compounds for biochemical treatment process of coal chemical wastewater in pilot scale[J]. Environ Pollut, 2023, 331(Pt 2): 121872. |
| [11] | Ge XY, Ma XF, Wu ZS, et al. Modification of coal-based activated carbon with nitric acid using microwave radiation for adsorption of phenanthrene and naphthalene[J]. Res Chem Intermed, 2015, 41(10): 7327-7347. |
| [12] | Mai ZM, Wang L, Li QQ, et al. Biodegradation and metabolic pathway of phenanthrene by a newly isolated bacterium Gordonia sp. SCSIO19801[J]. Biochem Biophys Res Commun, 2021, 585: 42-47. |
| [13] | Avramova T, Sotirova A, Galabova D, et al. Effect of Triton X-100 and rhamnolipid PS-17 on the mineralization of phenanthrene by Pseudomonas sp. cells[J]. Int Biodeterior Biodegrad, 2008, 62(4): 415-420. |
| [14] | 张莉, 徐智敏, 孙亚军, 等. 鄂尔多斯典型煤矿不同功能区水化学与微生物群落特征及环境响应[J]. 煤炭科学技术, 2023, 51(12):180-196. |
| Zhang L, Xu ZM, Sun YJ, et al. Hydrochemistry and microbial community characteristics and environmental response in different functional zones of a typical coal mine in Ordos[J]. Coal Science and Technology, 2023, 51(12): 180-196. | |
| [15] | 陈家玉, 桂和荣, 郭艳, 等. 淮北煤田深层地下水微生物群落特征及其水源示踪意义[J]. 煤炭学报, 2023, 48(9):3503-12. |
| Chen JY, Gui HR, Guo Y, et al. Microbial community characteristics of deep groundwater in Huaibel coalfield and its significance in water source tracing[J]. Joumnal of China Coal Society, 2023, 48(9):3503-12. | |
| [16] |
Mukherjee A, Reddy MS. Metatranscriptomics: an approach for retrieving novel eukaryotic genes from polluted and related environments[J]. 3 Biotech, 2020, 10(2): 71.
doi: 10.1007/s13205-020-2057-1 pmid: 32030340 |
| [17] | Jha P, Ghosh S, Vidyarthi AS, et al. Unravelling the microbial community structure and function of coal-bed methane producing formation water of Jharia coal mines using metagenomics approach[J]. Fuel, 2022, 317: 123459. |
| [18] | Liu BJ, Wang YW, Li Y, et al. Improved formation of biogenic methane by cultivable bacteria in highly volatile bituminous coals[J]. J Clean Prod, 2022, 366: 132900. |
| [19] | Guo HY, Jia WQ, Chen ZH, et al. Analysis on methane production from various coal slime fermentations based on metagenomics[J]. J Environ Manage, 2023, 343: 118058. |
| [20] | Niu SY, Yang JY, McDermaid A, et al. Bioinformatics tools for quantitative and functional metagenome and metatranscriptome data analysis in microbes[J]. Brief Bioinform, 2018, 19(6): 1415-1429. |
| [21] | Chung YW, Gwak HJ, Moon S, et al. Functional dynamics of bacterial species in the mouse gut microbiome revealed by metagenomic and metatranscriptomic analyses[J]. PLoS One, 2020, 15(1): e0227886. |
| [22] | Kamke J, Kittelmann S, Soni P, et al. Rumen metagenome and metatranscriptome analyses of low methane yield sheep reveals a Sharpea-enriched microbiome characterised by lactic acid formation and utilisation[J]. Microbiome, 2016, 4(1): 56. |
| [23] | Crovadore J, Soljan V, Calmin G, et al. Metatranscriptomic and metagenomic description of the bacterial nitrogen metabolism in waste water wet oxidation effluents[J]. Heliyon, 2017, 3(10): e00427. |
| [24] | Feng X, Zhang ZZ, Guo HG, et al. Enhancement of biogenic methane production from coal using supercritical CO2[J]. J Supercrit Fluids, 2023, 201: 106023. |
| [25] | Chen SF, Zhou YQ, Chen YR, et al. Fastp: an ultra-fast all-in-one FASTQ preprocessor[J]. Bioinformatics, 2018, 34(17): i884-i890. |
| [26] |
Li DH, Liu CM, Luo RB, et al. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph[J]. Bioinformatics, 2015, 31(10): 1674-1676.
doi: 10.1093/bioinformatics/btv033 pmid: 25609793 |
| [27] |
Fu LM, Niu BF, Zhu ZW, et al. CD-HIT: accelerated for clustering the next-generation sequencing data[J]. Bioinformatics, 2012, 28(23): 3150-3152.
doi: 10.1093/bioinformatics/bts565 pmid: 23060610 |
| [28] |
Li RQ, Li YR, Kristiansen K, et al. SOAP: short oligonucleotide alignment program[J]. Bioinformatics, 2008, 24(5): 713-714.
doi: 10.1093/bioinformatics/btn025 pmid: 18227114 |
| [29] |
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND[J]. Nat Methods, 2015, 12(1): 59-60.
doi: 10.1038/nmeth.3176 pmid: 25402007 |
| [30] |
Wu JY, Gao WM, Johnson RH, et al. Integrated metagenomic and metatranscriptomic analyses of microbial communities in the meso- and bathypelagic realm of North Pacific Ocean[J]. Mar Drugs, 2013, 11(10): 3777-3801.
doi: 10.3390/md11103777 pmid: 24152557 |
| [31] | Yang G, Yin YN, Wang JL. Microbial community diversity during fermentative hydrogen production inoculating various pretreated cultures[J]. Int J Hydrog Energy, 2019, 44(26): 13147-13156. |
| [32] | Li JB, Meng DL, Wang XZ, et al. Sources and succession of microorganisms in industrial coal flotation system[J]. Fuel, 2023, 342: 127917. |
| [33] | Ruan MY, Hu ZQ, Zhu Q, et al. 16S rDNA sequencing-based insights into the bacterial community structure and function in co-existing soil and coal gangue[J]. Microorganisms, 2023, 11(9): 2151. |
| [34] | Wang DX, Han HJ, Han YX, et al. Enhanced treatment of Fischer-Tropsch(F-T)wastewater using the up-flow anaerobic sludge blanket coupled with bioelectrochemical system: effect of electric field[J]. Bioresour Technol, 2017, 232: 18-26. |
| [35] | Li WW, Khalid H, Zhu Z, et al. Methane production through anaerobic digestion: participation and digestion characteristics of cellulose, hemicellulose and lignin[J]. Appl Energy, 2018, 226: 1219-1228. |
| [36] |
Laothanachareon T, Kanchanasuta S, Mhuanthong W, et al. Analysis of microbial community adaptation in mesophilic hydrogen fermentation from food waste by tagged 16S rRNA gene pyrosequencing[J]. J Environ Manage, 2014, 144: 143-151.
doi: 10.1016/j.jenvman.2014.05.019 pmid: 24945701 |
| [37] | Aüllo T, Berlendis S, Lascourrèges JF, et al. New bio-indicators for long term natural attenuation of monoaromatic compounds in deep terrestrial aquifers[J]. Front Microbiol, 2016, 7: 122. |
| [38] | Orem WH, Tatu CA, Lerch HE, et al. Organic compounds in produced waters from coalbed natural gas wells in the Powder River Basin, Wyoming, USA[J]. Appl Geochem, 2007, 22(10): 2240-2256. |
| [39] | Ulrich G, Bower S. Active methanogenesis and acetate utilization in Powder River Basin coals, United States[J]. Int J Coal Geol, 2008, 76(1/2): 25-33. |
| [40] | Zhu JG, Liu RY, Cao N, et al. Mycobacterial metabolic characteristics in a water meter biofilm revealed by metagenomics and metatranscriptomics[J]. Water Res, 2019, 153: 315-323. |
| [41] | Zhao HP, Liang SH, Yang XE. Isolation and characterization of catechol 2, 3-dioxygenase genes from phenanthrene degraders Sphingomonas sp. ZP1 and Pseudomonas sp. ZP2[J]. Environ Technol, 2011, 33(15-16): 1895-1901. |
| [42] | Grifoll M, Selifonov SA, Chapman PJ. Evidence for a novel pathway in the degradation of fluorene by Pseudomonas sp. strain F274[J]. Appl Environ Microbiol, 1994, 60(7): 2438-2449. |
| [43] | Lin Q, De Vrieze J, He GH, et al. Temperature regulates methane production through the function centralization of microbial community in anaerobic digestion[J]. Bioresour Technol, 2016, 216: 150-158. |
| [44] | Liang JS, Nabi M, Zhang PY, et al. Promising biological conversion of lignocellulosic biomass to renewable energy with rumen microorganisms: a comprehensive review[J]. Renew Sustain Energy Rev, 2020, 134: 110335. |
| [45] | Zhong YJ, He JG, Wu F, et al. Metagenomic analysis reveals the size effect of magnetite on anaerobic digestion of waste activated sludge after thermal hydrolysis pretreatment[J]. Sci Total Environ, 2022, 851(Pt 1): 158133. |
| [46] | Chen Q, Liu CQ, Liu XY, et al. Magnetite enhances anaerobic digestion of high salinity organic wastewater[J]. Environ Res, 2020, 189: 109884. |
| [1] | ZHANG Di, JU Rui, LI Li-mei, WANG Yu-qian, CHEN Rui, WANG Xin-yi. Application of Transcription Factor-based Biosensors in Environmental Analysis [J]. Biotechnology Bulletin, 2024, 40(6): 114-125. |
| [2] | LU Zhao-xiang, WANG Xi-ran, LIAN Xin-lei, LIAO Xiao-ping, LIU Ya-hong, SUN Jian. Advances in the Discovery of Novel Antibiotic-resistant Genes Based on Functional Metagenomics [J]. Biotechnology Bulletin, 2022, 38(9): 17-27. |
| [3] | MA Tao, LU Wei, LI Song-li, FAN Xia. Research Advance in the Resistome in the Microbiome of Livestock Animals [J]. Biotechnology Bulletin, 2021, 37(1): 113-122. |
| [4] | WANG Pan-pan, YANG Ye, LIU Di-qiu, CUI Xiu-ming, LIU Yuan. Application of Metagenomics in Plant Diseases Research [J]. Biotechnology Bulletin, 2020, 36(12): 146-154. |
| [5] | WANG Ye, JIA Zhen-hua, SONG Shui-shan. Research Advances on Integrating Metagenomics and Synthetic Biology in Discovering Novel Biocatalysts [J]. Biotechnology Bulletin, 2018, 34(8): 35-42. |
| [6] | LI Pei-han, LI Peng, SONG Hong-bin. Application of Metagenomics in Prevention and Control of Infectious Diseases [J]. Biotechnology Bulletin, 2018, 34(3): 43-52. |
| [7] | WANG Zhu-jun, WANG Shang, LIU Yang-ying, FENG Kai, DENG Ye. The Applications of Metagenomics in the Detection of Environmental Microbes Involving in Nitrogen Cycle [J]. Biotechnology Bulletin, 2018, 34(1): 1-14. |
| [8] | YAO Xue, LIU Wen-li, PEI Guang-qian, TONG Yi-gang, LUO Ya-ping. Optimization of Method for Analyzing Microbial Community on Human Skin by Targeted Sequencing of Metagenomics [J]. Biotechnology Bulletin, 2016, 32(11): 137-143. |
| [9] | Zhang Jun, Zhao Shengguo, Wang Jiaqi, Jin Di, Bu Dengpan. Research Progress of Enzyme and Relevant Gene Screened from Rumen by Metagenomic Approach [J]. Biotechnology Bulletin, 2015, 31(5): 32-40. |
| [10] | Liu Jiemeng, Qi Ji. Progress in the Study of Environmental Microbes by Metagenomic Methods [J]. Biotechnology Bulletin, 2015, 31(11): 51-59. |
| [11] | Mai Zhimao, Su Hongfei, Li Lizhen, Zhang Si. Construction of a Mangrove Soil Metagenome Library and Identification of Two Novel β-Glucosidase Genes [J]. Biotechnology Bulletin, 2014, 30(6): 168-172. |
| [12] | Xu Hao, Luo Xi, Li Yun, Xue Yang, Ye Qin. Applications of Environmental DNA Approaches to Ecological Researches [J]. Biotechnology Bulletin, 2014, 30(10): 49-55. |
| [13] | Liu Xinxing, Yun Hui, Xie Jianping, Huo Zhuanzhuan, Wu Haiyan, Yang Yingjie. Research Progress of Magnetosome Formation Genes and Proteins [J]. Biotechnology Bulletin, 2013, 0(8): 28-35. |
| [14] | Ma Shu, Liu Huhu, Tian Yun, Lu Xiangyang . Advances of Metatranscriptomics Technology [J]. Biotechnology Bulletin, 2012, 0(12): 46-50. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||