







Biotechnology Bulletin ›› 2025, Vol. 41 ›› Issue (12): 280-293.doi: 10.13560/j.cnki.biotech.bull.1985.2025-0452
Previous Articles Next Articles
SI Xu-peng1,2( ), CUI Xiu-wen1,2, OUYANG Li-zhi3, FANG Dan-dan1,2, PEI Long-ying1,2, FANG Gui-ping1,2, PU Xi-lei1,2, MA Yi-mian3, ZHANG Zheng3()
), CUI Xiu-wen1,2, OUYANG Li-zhi3, FANG Dan-dan1,2, PEI Long-ying1,2, FANG Gui-ping1,2, PU Xi-lei1,2, MA Yi-mian3, ZHANG Zheng3()
Received:2025-05-01
Online:2025-12-26
Published:2026-01-06
Contact:
ZHANG Zheng
E-mail:sixupeng0527@163.com;zhangzheng@implad.ac.cn
SI Xu-peng, CUI Xiu-wen, OUYANG Li-zhi, FANG Dan-dan, PEI Long-ying, FANG Gui-ping, PU Xi-lei, MA Yi-mian, ZHANG Zheng. Transcriptome Analysis of Different Parts of Ferula Sinkiangensis K. M. Shen and Exploration of Genes Related to Sesquiterpene Synthesis[J]. Biotechnology Bulletin, 2025, 41(12): 280-293.
基因名 Gene name | 基因ID Gene ID | 引物序列 Primer sequence (5′-3′) |
|---|---|---|
| P450 | Cluster-8529.5884-F | CGGTCCGAGAAGTTATGGGG |
| Cluster-8529.5884-R | TGGCAACCACCCTCTTCATC | |
| DXS | Cluster-8529.15434-F | GCTGGACATGGGTGTATGCT |
| Cluster-8529.15434-R | CTGGAGTTTTGCTCCCCCAT | |
| AACT | Cluster-8529.15178-F | CTGCGGTTAAATCTGCTGGT |
| Cluster-8529.15178-R | GTTACTCCCAAAGGATGCCCC | |
| TPS | Cluster-8529.5582-F | TACTGGATGATGAGGGAGCGA |
| Cluster-8529.5582-R | ACAATACGAGTGGAGCCGC | |
| HMGR | Cluster-8529.7778-F | TTCGAGACGCTTGCACTCAT |
| Cluster-8529.7778-R | TTGCATCCCCAGTGCTACAG | |
| DXR | Cluster-8529.20686-F | TGTCAGAAGCACCAAGACGA |
| Cluster-8529.20686-R | TACAAGAGCGGGACTCAAGC | |
| Actin | Actin-F | TGGTATTGTGCTGGATTCTGGT |
| Actin-R | TGAGATCACCACCAGCAAGG |
Table 1 Primer sequence for RT-qPCR
基因名 Gene name | 基因ID Gene ID | 引物序列 Primer sequence (5′-3′) |
|---|---|---|
| P450 | Cluster-8529.5884-F | CGGTCCGAGAAGTTATGGGG |
| Cluster-8529.5884-R | TGGCAACCACCCTCTTCATC | |
| DXS | Cluster-8529.15434-F | GCTGGACATGGGTGTATGCT |
| Cluster-8529.15434-R | CTGGAGTTTTGCTCCCCCAT | |
| AACT | Cluster-8529.15178-F | CTGCGGTTAAATCTGCTGGT |
| Cluster-8529.15178-R | GTTACTCCCAAAGGATGCCCC | |
| TPS | Cluster-8529.5582-F | TACTGGATGATGAGGGAGCGA |
| Cluster-8529.5582-R | ACAATACGAGTGGAGCCGC | |
| HMGR | Cluster-8529.7778-F | TTCGAGACGCTTGCACTCAT |
| Cluster-8529.7778-R | TTGCATCCCCAGTGCTACAG | |
| DXR | Cluster-8529.20686-F | TGTCAGAAGCACCAAGACGA |
| Cluster-8529.20686-R | TACAAGAGCGGGACTCAAGC | |
| Actin | Actin-F | TGGTATTGTGCTGGATTCTGGT |
| Actin-R | TGAGATCACCACCAGCAAGG |

Fig. 1 GO functional classification of Unigenes in F. sinkiangensis

Fig. 2 KOG annotation classification in the transcriptome of F. sinkiangensis
| 序号 Serial number | 通路ID Pathway ID | 代谢通路 Metabolic pathway | 数量 Quantity |
|---|---|---|---|
| 1 | ko00523 | Polyketide sugar unit biosynthesis | 3 |
| 2 | ko00900 | Terpenoid backbone biosynthesis | 59 |
| 3 | ko00902 | Monoterpenoid biosynthesis | 7 |
| 4 | ko00903 | Limonene degradation | 12 |
| 5 | ko00904 | Diterpenoid biosynthesis | 24 |
| 6 | ko00905 | Brassinosteroid biosynthesis | 18 |
| 7 | ko00906 | Carotenoid biosynthesis | 33 |
| 8 | ko00908 | Zeatin biosynthesis | 34 |
| 9 | ko00909 | Sesquiterpenoid and triterpenoid biosynthesis | 15 |
| 10 | ko01051 | Biosynthesis of ansamycins | 2 |
| 11 | ko01053 | Biosynthesis of siderophore group nonribosomal peptides | 1 |
Table 2 KEGG pathway analysis of terpenoids and polyketides
| 序号 Serial number | 通路ID Pathway ID | 代谢通路 Metabolic pathway | 数量 Quantity |
|---|---|---|---|
| 1 | ko00523 | Polyketide sugar unit biosynthesis | 3 |
| 2 | ko00900 | Terpenoid backbone biosynthesis | 59 |
| 3 | ko00902 | Monoterpenoid biosynthesis | 7 |
| 4 | ko00903 | Limonene degradation | 12 |
| 5 | ko00904 | Diterpenoid biosynthesis | 24 |
| 6 | ko00905 | Brassinosteroid biosynthesis | 18 |
| 7 | ko00906 | Carotenoid biosynthesis | 33 |
| 8 | ko00908 | Zeatin biosynthesis | 34 |
| 9 | ko00909 | Sesquiterpenoid and triterpenoid biosynthesis | 15 |
| 10 | ko01051 | Biosynthesis of ansamycins | 2 |
| 11 | ko01053 | Biosynthesis of siderophore group nonribosomal peptides | 1 |

Fig. 4 Pathway closely related to the formation of pharmacological substancesA: Biosynthesis of other secondary metabolites. B: Metabolism of terpenoids and polyketides
| 骨架 Skeleton | 名称 Name | 数量 Quantity | FPKM (Fragments Per) |
|---|---|---|---|
萜类骨架 Terpenoid skeleton(59)ko00900 | Geranylgeranyl pyrophosphate synthase | 3 | 6.62-14.69 |
| Farnesol kinase | 1 | 4.79-7.41 | |
| 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (MDS) | 1 | 5.3-6.11 | |
| 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (MCT) | 3 | 10.67-29.75 | |
| Prenyl protein peptidase | 1 | 3.66-6.47 | |
| Diphosphomevalonate decarboxylase (MVD) | 2 | 4.33-10.76 | |
| Hydroxymethylglutaryl-CoA synthase (HMGCS) | 2 | 6.53-37.72 | |
| Farnesylcysteine lyase | 1 | 39-45.25 | |
| Geranylgeranyl diphosphate reductase | 2 | 3.17-46.74 | |
| Acetyl-CoA C-acetyltransferase (ACAT) | 1 | 23.22-41.51 | |
| Polycis-polyprenyl diphosphate synthase | 2 | 2.29-22.71 | |
| Geranyl diphosphate synthase (GPS) | 3 | 0.39-14.17 | |
| 4-Hydroxy-3-methylbut-2-enyl diphosphate reductase (HDR) | 5 | 0.1-25.06 | |
| Hypothetical protein | 1 | 0.38-1.52 | |
| 1-Deoxy-D-xylulose-5-phosphate synthase (DXS) | 3 | 3.58-43.29 | |
| 3-Hydroxy-3-methylglutaryl-coenzyme A reductase (HMGR) | 3 | 0.43-60.75 | |
| 4-Hydroxy-3-methylbut-2-enyl-diphosphate synthase (HXS) | 1 | 25.93-42.74 | |
| CAAX prenyl protease | 1 | 60.08-65.9 | |
| Mevalonate kinase (MK) | 1 | 16.24-18.02 | |
| Phosphomevalonate kinase (MVK2) | 1 | 8.52-11.37 | |
| Isopentenyl-diphosphate Delta-isomerase | 1 | 75.21-102.72 | |
| Acetyl-CoA C-acetyltransferase | 4 | 0.16-83.75 | |
| Protein-S-isoprenylcysteine O-methyltransferase (STE14) | 1 | 10.42-17.69 | |
| Isopentenyl phosphate kinase | 1 | 8.28-10.05 | |
| Farnesyl diphosphate synthase (FDPS) | 1 | 5.67-7.64 | |
| Dihydroflavonol-4-reductase | 3 | 0.42-18.19 | |
| Prenylcysteine alpha-carboxyl methylesterase | 1 | 3.53-4.22 | |
| Protein farnesyltransferase/geranylgeranyltransferase | 1 | 13.1-14.01 | |
| Geranylgeranyl pyrophosphate synthase/Polyprenyl synthetase | 1 | 6.66-8.04 | |
| 4-Diphosphocytidyl-2-C-methyl-D-erythritol kinase (CMK) | 1 | 12.65-15.84 | |
| Solanesyl-diphosphate synthase | 1 | 2.35-3.24 | |
| Protein farnesyltransferase subunit beta (FNTB) | 1 | 8.47-9.76 | |
| Ditrans,polycis-polyprenyl diphosphate synthase | 1 | 11.54-14.64 | |
| Prenyl protease | 1 | 12.7-13.94 | |
倍半萜及三萜生物合成 Biosynthesis of sesquiterpenes and triterpenes(15)ko00909 | Sesquiterpene synthase | 3 | 0.33-10.01 |
| Beta-amyrin synthase | 2 | 1.05-3.53 | |
| Squalene epoxidase | 4 | 0.14-32.06 | |
| Dihydroflavonol-4-reductase | 3 | 0.16-18.19 | |
| Squalene synthase | 2 | 0.42-58.1 | |
| (3S, 6E)-Nerolidol synthase | 1 | 1.97-3.91 |
Table 3 Unigenes related to terpenoid backbone and sesquiterpene synthesis in F. sinkiangensis
| 骨架 Skeleton | 名称 Name | 数量 Quantity | FPKM (Fragments Per) |
|---|---|---|---|
萜类骨架 Terpenoid skeleton(59)ko00900 | Geranylgeranyl pyrophosphate synthase | 3 | 6.62-14.69 |
| Farnesol kinase | 1 | 4.79-7.41 | |
| 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (MDS) | 1 | 5.3-6.11 | |
| 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (MCT) | 3 | 10.67-29.75 | |
| Prenyl protein peptidase | 1 | 3.66-6.47 | |
| Diphosphomevalonate decarboxylase (MVD) | 2 | 4.33-10.76 | |
| Hydroxymethylglutaryl-CoA synthase (HMGCS) | 2 | 6.53-37.72 | |
| Farnesylcysteine lyase | 1 | 39-45.25 | |
| Geranylgeranyl diphosphate reductase | 2 | 3.17-46.74 | |
| Acetyl-CoA C-acetyltransferase (ACAT) | 1 | 23.22-41.51 | |
| Polycis-polyprenyl diphosphate synthase | 2 | 2.29-22.71 | |
| Geranyl diphosphate synthase (GPS) | 3 | 0.39-14.17 | |
| 4-Hydroxy-3-methylbut-2-enyl diphosphate reductase (HDR) | 5 | 0.1-25.06 | |
| Hypothetical protein | 1 | 0.38-1.52 | |
| 1-Deoxy-D-xylulose-5-phosphate synthase (DXS) | 3 | 3.58-43.29 | |
| 3-Hydroxy-3-methylglutaryl-coenzyme A reductase (HMGR) | 3 | 0.43-60.75 | |
| 4-Hydroxy-3-methylbut-2-enyl-diphosphate synthase (HXS) | 1 | 25.93-42.74 | |
| CAAX prenyl protease | 1 | 60.08-65.9 | |
| Mevalonate kinase (MK) | 1 | 16.24-18.02 | |
| Phosphomevalonate kinase (MVK2) | 1 | 8.52-11.37 | |
| Isopentenyl-diphosphate Delta-isomerase | 1 | 75.21-102.72 | |
| Acetyl-CoA C-acetyltransferase | 4 | 0.16-83.75 | |
| Protein-S-isoprenylcysteine O-methyltransferase (STE14) | 1 | 10.42-17.69 | |
| Isopentenyl phosphate kinase | 1 | 8.28-10.05 | |
| Farnesyl diphosphate synthase (FDPS) | 1 | 5.67-7.64 | |
| Dihydroflavonol-4-reductase | 3 | 0.42-18.19 | |
| Prenylcysteine alpha-carboxyl methylesterase | 1 | 3.53-4.22 | |
| Protein farnesyltransferase/geranylgeranyltransferase | 1 | 13.1-14.01 | |
| Geranylgeranyl pyrophosphate synthase/Polyprenyl synthetase | 1 | 6.66-8.04 | |
| 4-Diphosphocytidyl-2-C-methyl-D-erythritol kinase (CMK) | 1 | 12.65-15.84 | |
| Solanesyl-diphosphate synthase | 1 | 2.35-3.24 | |
| Protein farnesyltransferase subunit beta (FNTB) | 1 | 8.47-9.76 | |
| Ditrans,polycis-polyprenyl diphosphate synthase | 1 | 11.54-14.64 | |
| Prenyl protease | 1 | 12.7-13.94 | |
倍半萜及三萜生物合成 Biosynthesis of sesquiterpenes and triterpenes(15)ko00909 | Sesquiterpene synthase | 3 | 0.33-10.01 |
| Beta-amyrin synthase | 2 | 1.05-3.53 | |
| Squalene epoxidase | 4 | 0.14-32.06 | |
| Dihydroflavonol-4-reductase | 3 | 0.16-18.19 | |
| Squalene synthase | 2 | 0.42-58.1 | |
| (3S, 6E)-Nerolidol synthase | 1 | 1.97-3.91 |
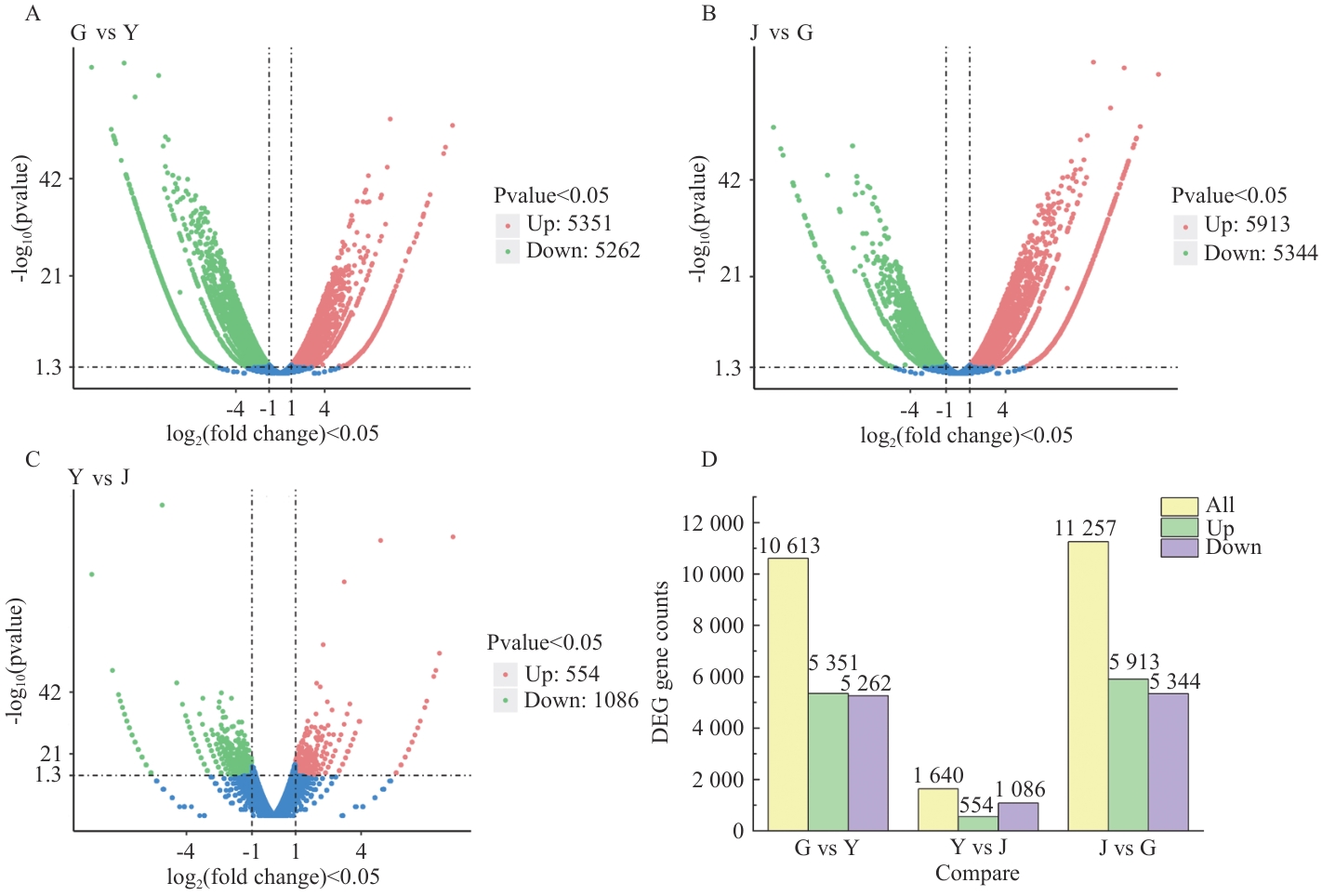
Fig. 5 Distribution and expression of differentially expressed genes in different parts of F. sinkiangensisA-C: Distribution of differentially expressed genes. D: Expression of differential genes
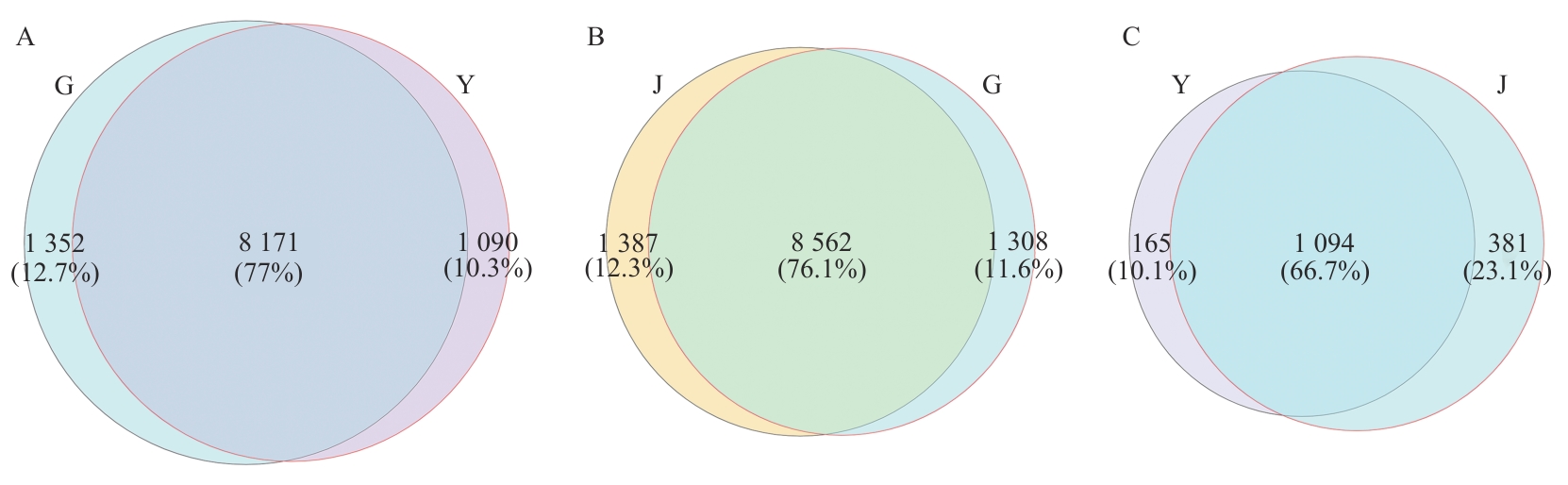
Fig. 6 Venn diagram of comparing the combined differential genesG: Root; Y: leaf; J: Stem. The same below
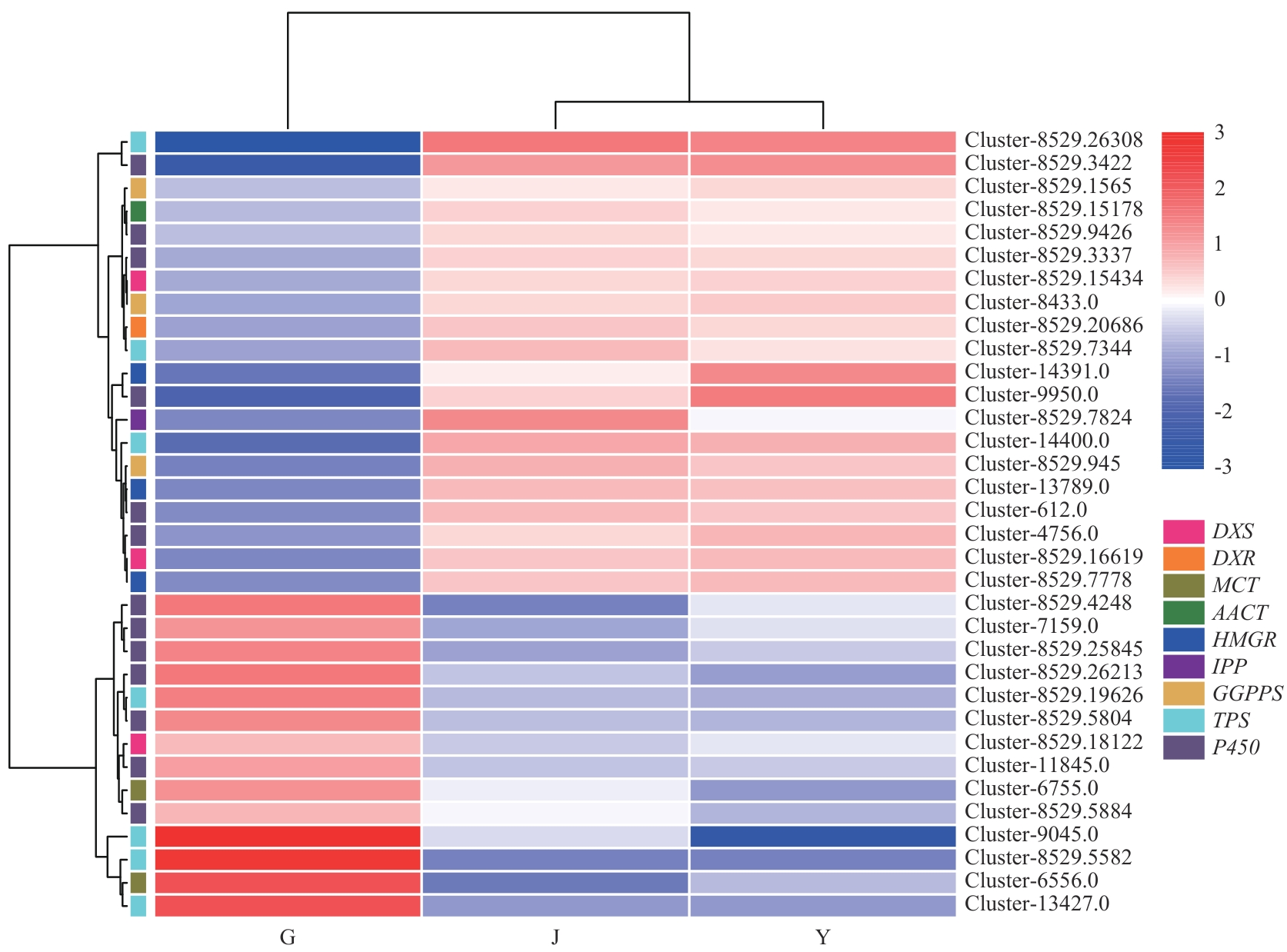
Fig. 7 Clustering heat map of differential metabolic genes
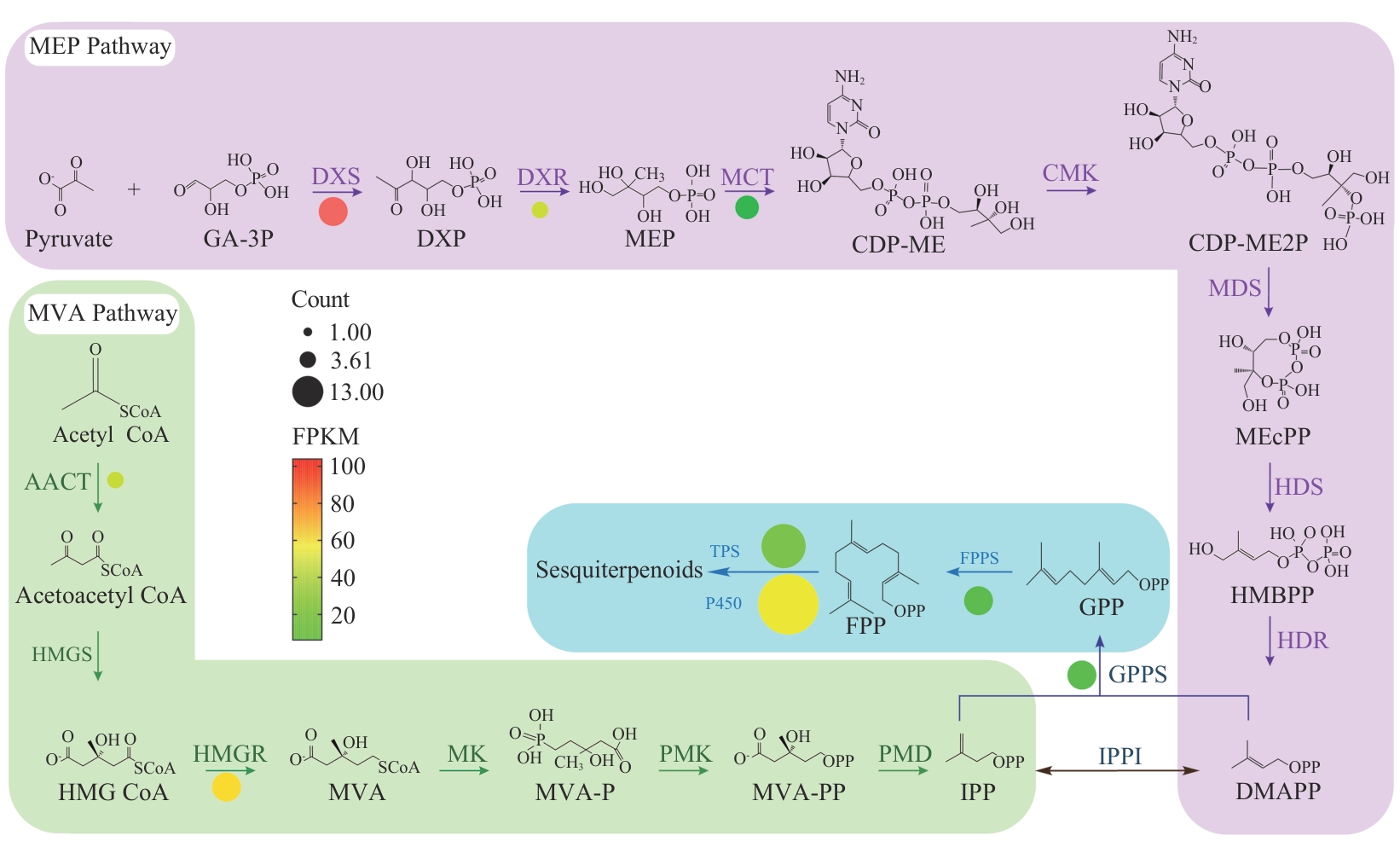
Fig. 8 Biosynthetic pathway of sesquiterpenes in F. sinkiangensis
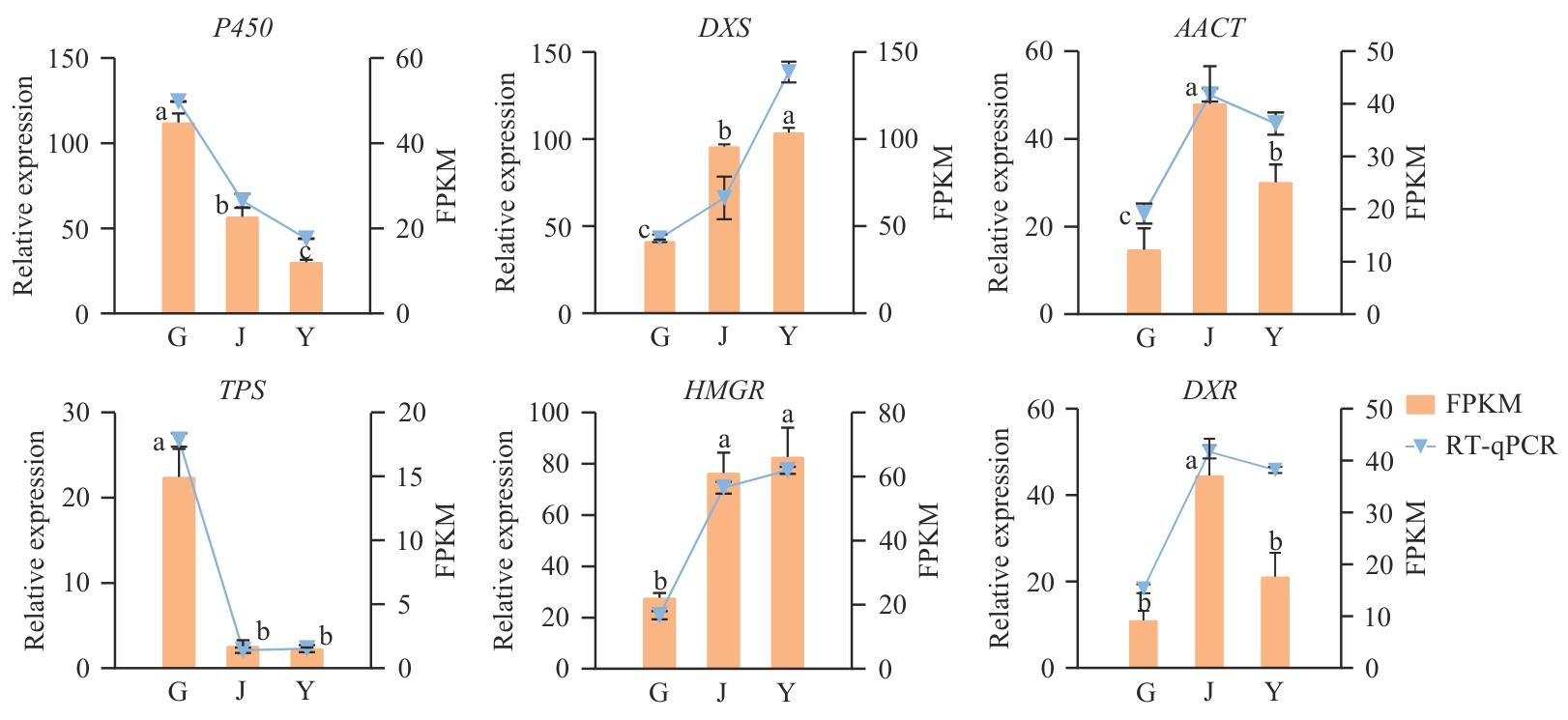
Fig. 9 Verification of gene expressions related to sesquiterpene synthesis in different parts of F. sinkiangensisData with different lowercase letters indicate significant difference at 0.05 level
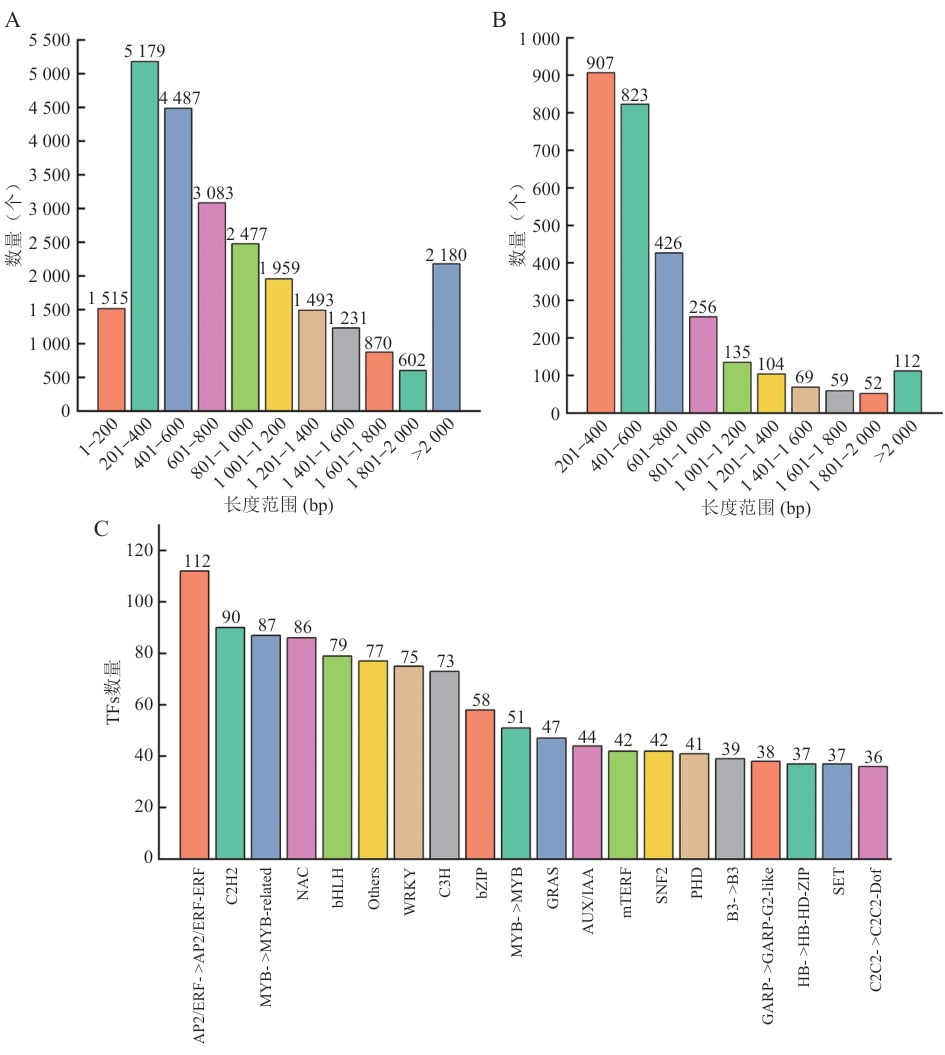
Fig. 10 Distribution of CDS lengths and number of transcription factor familyA: Blast prediction. B: TransDecoder prediction. C: Number of TFs
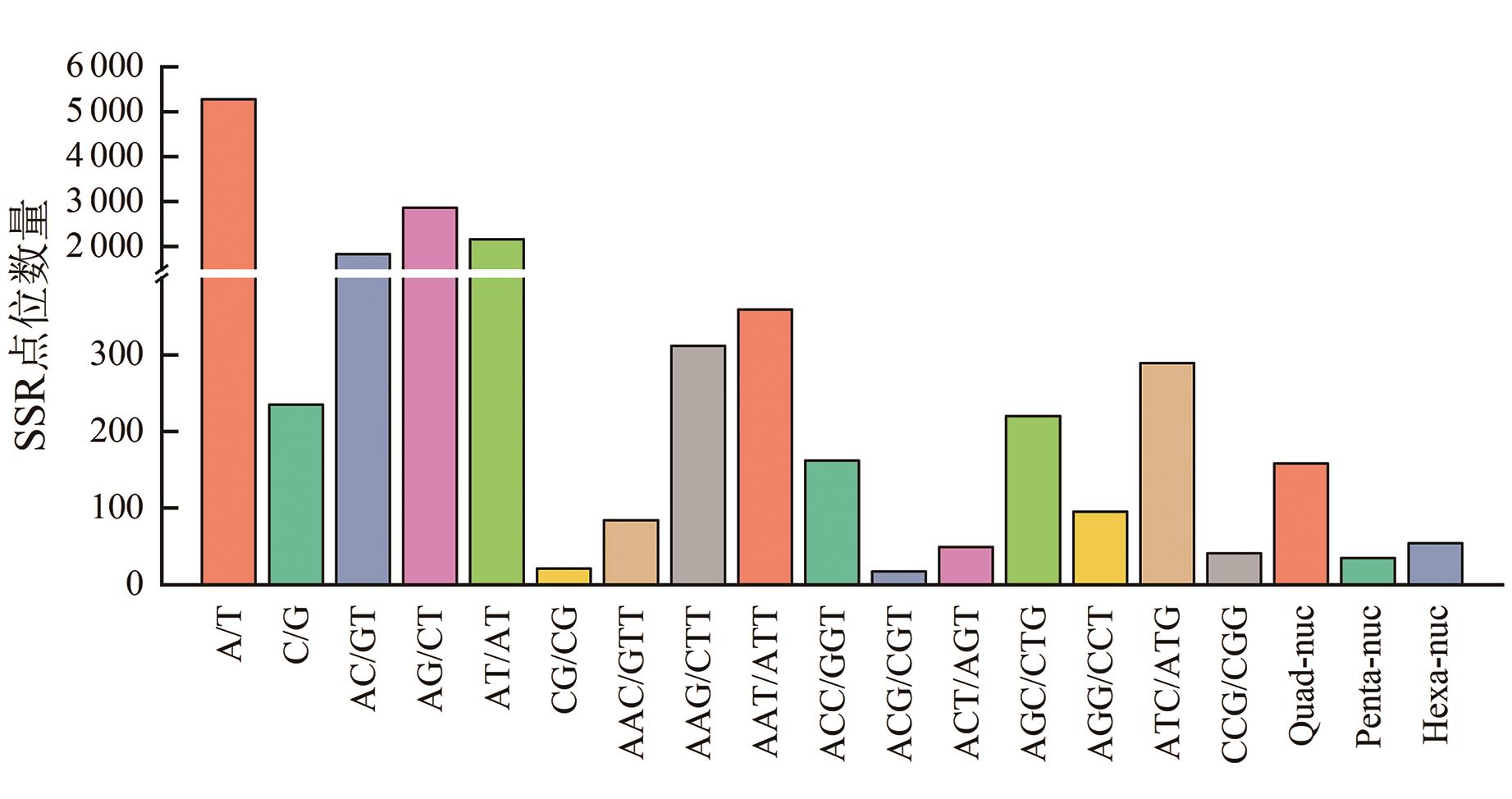
Fig. 11 Distribution of nucleotide motifs in SSR
重复基序数目 Number of repeated motifs | SSR位点数目 Number of SSR loci | 总数量 Total quantity | |||||
|---|---|---|---|---|---|---|---|
单核苷酸 Single nucleotide | 二核苷酸 Dinucleotide | 三核苷酸 Trinucleotide | 四核苷酸 Tetranucleotide | 五核苷酸 Pentanucleotide | 六核苷酸 Hexanucleotide | ||
| 5 | 0 | 0 | 844 | 96 | 27 | 41 | 1 008 |
| 6 | 0 | 1800 | 371 | 36 | 8 | 5 | 2 220 |
| 7 | 0 | 1208 | 181 | 18 | 0 | 4 | 1 411 |
| 8 | 0 | 939 | 83 | 5 | 0 | 3 | 1 030 |
| 9 | 0 | 757 | 60 | 1 | 0 | 0 | 818 |
| 10 | 5 | 534 | 42 | 0 | 0 | 0 | 581 |
| 11 | 6 | 338 | 3 | 0 | 0 | 0 | 347 |
| 12 | 7 | 280 | 9 | 1 | 0 | 0 | 297 |
| 13 | 8 | 202 | 9 | 0 | 0 | 0 | 219 |
| 14 | 9 | 227 | 6 | 0 | 0 | 0 | 242 |
| 15 | 15 | 153 | 5 | 1 | 0 | 0 | 174 |
| 16 | 17 | 66 | 1 | 0 | 0 | 0 | 84 |
| 17 | 19 | 72 | 1 | 0 | 0 | 1 | 93 |
| 18 | 21 | 86 | 3 | 0 | 0 | 0 | 110 |
| 19 | 23 | 51 | 2 | 0 | 0 | 0 | 76 |
| 20 | 30 | 54 | 2 | 0 | 0 | 0 | 86 |
| >20 | 198 | 125 | 6 | 0 | 0 | 0 | 329 |
| 总数量 | 358 | 6 892 | 1 628 | 158 | 35 | 54 | 9 125 |
Table 4 Distribution of SSR repeat motif
重复基序数目 Number of repeated motifs | SSR位点数目 Number of SSR loci | 总数量 Total quantity | |||||
|---|---|---|---|---|---|---|---|
单核苷酸 Single nucleotide | 二核苷酸 Dinucleotide | 三核苷酸 Trinucleotide | 四核苷酸 Tetranucleotide | 五核苷酸 Pentanucleotide | 六核苷酸 Hexanucleotide | ||
| 5 | 0 | 0 | 844 | 96 | 27 | 41 | 1 008 |
| 6 | 0 | 1800 | 371 | 36 | 8 | 5 | 2 220 |
| 7 | 0 | 1208 | 181 | 18 | 0 | 4 | 1 411 |
| 8 | 0 | 939 | 83 | 5 | 0 | 3 | 1 030 |
| 9 | 0 | 757 | 60 | 1 | 0 | 0 | 818 |
| 10 | 5 | 534 | 42 | 0 | 0 | 0 | 581 |
| 11 | 6 | 338 | 3 | 0 | 0 | 0 | 347 |
| 12 | 7 | 280 | 9 | 1 | 0 | 0 | 297 |
| 13 | 8 | 202 | 9 | 0 | 0 | 0 | 219 |
| 14 | 9 | 227 | 6 | 0 | 0 | 0 | 242 |
| 15 | 15 | 153 | 5 | 1 | 0 | 0 | 174 |
| 16 | 17 | 66 | 1 | 0 | 0 | 0 | 84 |
| 17 | 19 | 72 | 1 | 0 | 0 | 1 | 93 |
| 18 | 21 | 86 | 3 | 0 | 0 | 0 | 110 |
| 19 | 23 | 51 | 2 | 0 | 0 | 0 | 76 |
| 20 | 30 | 54 | 2 | 0 | 0 | 0 | 86 |
| >20 | 198 | 125 | 6 | 0 | 0 | 0 | 329 |
| 总数量 | 358 | 6 892 | 1 628 | 158 | 35 | 54 | 9 125 |
| [1] | Yang JR, An Z, Li ZH, et al. Sesquiterpene coumarins from the roots of Ferula sinkiangensis and Ferula teterrima [J]. Chem Pharm Bull, 2006, 54(11): 1595-1598. |
| [2] | Xing YC, Li N, Zhou D, et al. Sesquiterpene coumarins from Ferula sinkiangensis act as neuroinflammation inhibitors [J]. Planta Med, 2017, 83(1/02): 135-142. |
| [3] | Li GZ, Li XJ, Cao L, et al. Steroidal esters from Ferula sinkiangensis [J]. Fitoterapia, 2014, 97: 247-252. |
| [4] | Ye BG, Wang S, Zhang L. Studies on the detoxification effects and acute toxicity of a mixture of Cis-sec-butyl-1-propoenyl disulphide and trans-sec-butyl-1-propoenyl disulphide isolated from crude essential oil of Ferula sinkiangensis K.M. Shen, a Chinese traditional herbal medicine [J]. Nat Prod Res, 2011, 25(12): 1161-1170. |
| [5] | Zhou YT, Xin F, Zhang GQ, et al. Recent advances on bioactive constituents in Ferula [J]. Drug Dev Res, 2017, 78(7): 321-331. |
| [6] | Bao XH, Li YP, Li Q, et al. Racemic norneolignans from the resin of Ferula sinkiangensis and their COX-2 inhibitory activity [J]. Fitoterapia, 2023, 164: 105341. |
| [7] | 李洁, 徐海燕, 贾晓光, 等. 阜康阿魏的化学成分及其生物活性研究进展 [J]. 新疆医科大学学报, 2012, 35(9): 1159-1161. |
| Li J, Xu HY, Jia XG, et al. Research progress on chemical constituents and biological activities of Ferula Fukang [J]. J Xinjiang Med Univ, 2012, 35(9): 1159-1161. | |
| [8] | Wang JC, Zheng Q, Wang HX, et al. Sesquiterpenes and sesquiterpene derivatives from Ferula: their chemical structures, biosynthetic pathways, and biological properties [J]. Antioxidants, 2024, 13(1): 7. |
| [9] | Khayat MT, Alharbi M, Ghazawi KF, et al. Ferula sinkiangensis (Chou-AWei, Chinese Ferula): traditional uses, phytoconstituents, biosynthesis, and pharmacological activities [J]. Plants, 2023, 12(4): 902. |
| [10] | Macrì R, Bava I, Scarano F, et al. In vitro evaluation of ferutinin Rich-Ferula communis L., ssp. glauca, root extract on Doxorubicin-induced cardiotoxicity: antioxidant properties and cell cycle modulation [J]. Int J Mol Sci, 2023, 24(16): 12735. |
| [11] | Mi Y, Jiao K, Xu JK, et al. Kellerin from Ferula sinkiangensis exerts neuroprotective effects after focal cerebral ischemia in rats by inhibiting microglia-mediated inflammatory responses [J]. J Ethnopharmacol, 2021, 269: 113718. |
| [12] | Arjmand Z, Hamburger M, Dastan D. Isolation and purification of terpenoid compounds from Ferula haussknechtii and evaluation of their antibacterial effects [J]. Nat Prod Res, 2023, 37(10): 1617-1624. |
| [13] | Wang JL, Sang CY, Wang J, et al. Sesquiterpene coumarins from Ferula sinkiangensis and their anti-pancreatic cancer effects [J]. Phytochemistry, 2023, 214: 113824. |
| [14] | Li GZ, Wang JC, Li XJ, et al. An unusual sesquiterpene coumarin from the seeds of Ferula sinkiangensis [J]. J Asian Nat Prod Res, 2016, 18(9): 891-896. |
| [15] | 樊丛照. 基于多组学分析的新疆阿魏开花调控机制研究 [D]. 北京: 北京协和医学院, 2024. |
| Fan CZ. Mechanism of flowering regulation in Ferula sinkiangensis based on multi-omies analysis [D]. Beijing: Peking Union Medical College, 2024. | |
| [16] | Tang XH, Li TJ, Hao ZY, et al. Identification of key genes involved in sesquiterpene synthesis in Nardostachys jatamansi based on transcriptome and component analysis [J]. Genes, 2024, 15(12): 1539. |
| [17] | Wang JC, Wang HJ, Zhang M, et al. Sesquiterpene coumarins from Ferula sinkiangensis K.M.Shen and their cytotoxic activities [J]. Phytochemistry, 2020, 180: 112531. |
| [18] | Wang JL, Zhao YM, Qiang Y. Chemical constituents from Ferula sinkiangensis and their chemotaxonomic significance [J]. Biochem Syst Ecol, 2022, 105: 104519. |
| [19] | Ahmed S, Zhan CS, Yang YY, et al. The transcript profile of a traditional Chinese medicine, Atractylodes lancea, revealing its sesquiterpenoid biosynthesis of the major active components [J]. PLoS One, 2016, 11(3): e0151975. |
| [20] | Vranová E, Coman D, Gruissem W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis [J]. Annu Rev Plant Biol, 2013, 64: 665-700. |
| [21] | 陈建, 赵德刚. 植物萜类生物合成相关酶类及其编码基因的研究进展 [J]. 分子植物育种, 2004, 2(6): 757-764. |
| Chen J, Zhao DG. Research advances on the enzymes and their coding gene involved in plant terpene biosynthesis [J]. Mol Plant Breed, 2004, 2(6): 757-764. | |
| [22] | 郭连安, 潘媛, 谭均, 等. 不同生长年限木香转录组分析及倍半萜合成基因挖掘 [J]. 西南农业学报, 2024, 37(9): 2042-2050. |
| Guo LA, Pan Y, Tan J, et al. Transcriptome analysis of Aucklandiae lappa from different growth years and mining genes related to sesquiterpenes synthesis [J]. Southwest China J Agric Sci, 2024, 37(9): 2042-2050. | |
| [23] | Kim BR, Kim SU, Chang YJ. Differential expression of three 1-deoxy-D-xylulose-5-phosphate synthase genes in rice [J]. Biotechnol Lett, 2005, 27(14): 997-1001. |
| [24] | 曾珊珊, 高婷, 谷梦雅, 等. 基于黄棪(Camellia sinensis)基因组的DXS基因家族鉴定及表达 [J]. 应用与环境生物学报, 2024, 30(4): 811-817. |
| Zeng SS, Gao T, Gu MY, et al. Genome-wide identification and expression of the DXS gene family in Huangdan (Camellia sinensis) [J]. Chin J Appl Environ Biol, 2024, 30(4): 811-817. | |
| [25] | Zhang XW, Guo JW, Cheng FY, et al. Cytochrome P450 enzymes in fungal natural product biosynthesis [J]. Nat Prod Rep, 2021, 38(6): 1072-1099. |
| [26] | 高世玺, 戎梅, 彭俊祥, 等. 细胞色素P450在植物倍半萜类化合物生物合成中的作用研究进展 [J]. 药学学报, 2024, 59(2): 313-321. |
| Gao SX, Rong M, Peng JX, et al. Research progress on the role of cytochrome P450 in plant sesquiterpene biosynthesis [J]. Acta Pharm Sin, 2024, 59(2): 313-321. | |
| [27] | Chen DH, Liu CJ, Ye HC, et al. Ri-mediated transformation of Artemisia annua with a recombinant farnesyl diphosphate synthase gene for artemisinin production [J]. Plant Cell Tissue Organ Cult, 1999, 57(3): 157-162. |
| [28] | Ichinose H, Kitaoka T. Insight into metabolic diversity of the brown-rot basidiomycete Postia placenta responsible for sesquiterpene biosynthesis: semi-comprehensive screening of cytochrome P450 monooxygenase involved in protoilludene metabolism [J]. Microb Biotechnol, 2018, 11(5): 952-965. |
| [29] | Shoji T, Yuan L. ERF gene clusters: working together to regulate metabolism [J]. Trends Plant Sci, 2021, 26(1): 23-32. |
| [30] | Liu YH, Khan AR, Gan YB. C2H2 zinc finger proteins response to abiotic stress in plants [J]. Int J Mol Sci, 2022, 23(5): 2730. |
| [31] | 李超颖. 新疆阿魏化学成分及高效液相指纹图谱研究 [D]. 乌鲁木齐: 新疆大学, 2014. |
| Li CY. Studies of the chemical constituents and the HPLC fingerprints of Ferula sinkiangensis K.M.Shen [D]. Urumqi: Xinjiang University, 2014. | |
| [32] | 田再民, 张立平, 孙庆林, 等. 小麦数量性状基因定位研究进展 [J]. 内蒙古农业科技, 2007, 35(3): 68-71. |
| Tian ZM, Zhang LP, Sun QL, et al. Research review on wheat QTLs [J]. Inn Mong Agric Sci Technol, 2007, 35(3): 68-71. | |
| [33] | Fan CJ, Liu QY, Zeng BS, et al. Development of simple sequence repeat (SSR) markers and genetic diversity analysis in blackwood (Acacia melanoxylon) clones in China [J]. Silvae Genet, 2016, 65(1): 49-54. |
| [34] | Cregan PB, Akkaya MS, Bhagwat AA, et al. Length polymorphisms of simple sequence repeat (SSR) DNA as molecular markers in plants [M]// United Kingdom: CRC Press, Plant genome analysis, 1994: 47-56. |
| [35] | Kaur S, Pembleton LW, Cogan NO, et al. Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers [J]. BMC Genom, 2012, 13(1): 104. |
| [36] | Yang QW, Jiang YJ, Wang YP, et al. SSR loci analysis in transcriptome and molecular marker development in Polygonatum sibiricum . [J]. BioMed Res Int, 2022, 2022(1): 4237913. |
| [37] | Liu YL, Zhang PF, Song ML, et al. Transcriptome analysis and development of SSR molecular markers in Glycyrrhiza uralensis fisch [J]. PLoS One, 2015, 10(11): e0143017. |
| [1] | LIU Jian-guo, LIU Ge-er, GUO Ying-xin, WANG Bin, WANG Yu-kun, LU Jin-feng, HUANG Wen-ting, ZHU Yun-na. Integrate Transcriptomic and Metabolomic Analysis of Fruits Quality Differences between ‘Guiyou No. 1’ and ‘Shatianyou’ Pomelo (Citrus maxima) [J]. Biotechnology Bulletin, 2025, 41(9): 168-181. |
| [2] | LIU Ze-zhou, DUAN Nai-bin, YUE Li-xin, WANG Qing-hua, YAO Xing-hao, GAO Li-min, KONG Su-ping. Analysis of Wax Components and Screening of Wax-deficient Gene Ggl-1 in Garlic (Allium sativum L.) [J]. Biotechnology Bulletin, 2025, 41(9): 219-231. |
| [3] | YAN Meng-yang, LIANG Xiao-yang, DAI Jun-ang, ZHANG Yan, GUAN Tuan, ZHANG Hui, LIU Liang-bo, SUN Zhi-hua. Screening of Amoxicillin-degrading Bacteria and Study on Its Degradation Mechanisms [J]. Biotechnology Bulletin, 2025, 41(9): 314-325. |
| [4] | LU Yao, YUAN Ping-ping, JIN Xin, MAO Xiang-hong, FAN Xiang-bin, BAI Xiao-dong. Genetic Diversity Analysis and Fingerprinting of Wild Potato and Landraces Based on SSR Markers [J]. Biotechnology Bulletin, 2025, 41(9): 94-104. |
| [5] | PEI Hong-xia, WANG Lu-yao, LI Sheng-mei, GAO Jing-xia. Genetic Diversity of 220 Pepper Germplasm Resources Using SCoT, SRAP, and SSR Molecular Markers [J]. Biotechnology Bulletin, 2025, 41(8): 165-174. |
| [6] | BAI Yu-guo, LI Wan-di, LIANG Jian-ping, SHI Zhi-yong, LU Geng-long, LIU Hong-jun, NIU Jing-ping. Growth-promoting Mechanism of Trichoderma harzianum T9131 on Astragalus membranaceus Seedlings [J]. Biotechnology Bulletin, 2025, 41(8): 175-185. |
| [7] | WANG Yue-chen, HAN Xin-qi, WEI Wen-min, CUI Zhao-lan, LUO Yang-mei, CHEN Peng-ru, WANG Hai-gang, LIU long-long, ZHANG Li, WANG Lun. Biological Basis Study for Grain Shattering in Proso Millet and Identification of Genes Regulating Grain Shattering [J]. Biotechnology Bulletin, 2025, 41(7): 164-171. |
| [8] | ZHANG Yue, BI Yu, MU Xue-nan, ZHENG Zi-wei, WANG Zhi-gang, XU Wei-hui. Biocontrol Characteristics of Strain JB7 against Fusarium graminearum [J]. Biotechnology Bulletin, 2025, 41(7): 261-271. |
| [9] | DUAN Min-jie, LI Yi-fei, WANG Chun-ping, HUANG Ren-zhong, HUANG Qi-zhong, ZHANG Shi-cai. Association Analysis and Fingerprint Map Construction of Fruit Color Traits in Pepper via SSR Markers [J]. Biotechnology Bulletin, 2025, 41(7): 81-94. |
| [10] | LI Cheng-hua, DOU Fei-fei, REN Yu-zhao, LIU Cai-xia, LIU Feng-lou, WANG Zhang-jun, LI Qing-feng. Effect of Exogenous Salicylic Acid on Wheat Infested with Blumeria graminis f. sp. tritici and Its Transcriptome Analysis [J]. Biotechnology Bulletin, 2025, 41(7): 272-280. |
| [11] | GUO Xiu-juan, FENG Yu, WU Rui-xiang, WANG Li-qin, YANG Jian-chun. Transcriptome Analysis of the Effect of Ca 2+ Treatment on the Seed Germination of Flax [J]. Biotechnology Bulletin, 2025, 41(7): 139-149. |
| [12] | DUAN Yong-hong, YANG Xin, YU Guan-qun, XIA Jun-jun, SONG Lu-shuai, BAI Xiao-dong, PENG Suo-tang. Genetic Diversity and Principal Component Analysis of 125 Potato Germplasm Resources [J]. Biotechnology Bulletin, 2025, 41(6): 130-143. |
| [13] | LIU Yuan, ZHAO Ran, LU Zhen-fang, LI Rui-li. Research Progress in the Biological Metabolic Pathway and Functions of Plant Carotenoids [J]. Biotechnology Bulletin, 2025, 41(5): 23-31. |
| [14] | HU Ruo-qun, ZENG Jing-jing, LIANG Wan-feng, CAO Jia-yu, HUANG Xiao-wei, LIANG Xiao-ying, QIU Ming-yue, CHEN Ying. Integrated Transcriptome and Metabolome Analysis to Explore the Carotenoid Synthesis and Metabolism Mechanism in Anoectochilus roxburghii under Different Shading Conditions [J]. Biotechnology Bulletin, 2025, 41(5): 231-243. |
| [15] | TANG You, ZHAO Jun-wei, SUN Lan-xi, LI Xiang. Combined Application of Multi-omics Technologies in Plant Metabolic Pathway Resolution [J]. Biotechnology Bulletin, 2025, 41(4): 76-87. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||