







Biotechnology Bulletin ›› 2026, Vol. 42 ›› Issue (6): 279-293.doi: 10.13560/j.cnki.biotech.bull.1985.2025-0927
WANG Jia-bin1( ), HU Yue2, CHEN Jia-jie1, WANG Meng2, HE Xiu-yun1, LI Zhi-yong2, LI Jian2, WANG Mei-na2()
), HU Yue2, CHEN Jia-jie1, WANG Meng2, HE Xiu-yun1, LI Zhi-yong2, LI Jian2, WANG Mei-na2()
Received:2025-08-27
Online:2026-06-26
Published:2026-07-11
Contact:
WANG Mei-na
E-mail:44376283@qq.com;snow-wmn2005@163.com
WANG Jia-bin, HU Yue, CHEN Jia-jie, WANG Meng, HE Xiu-yun, LI Zhi-yong, LI Jian, WANG Mei-na. Combined Metabolomic and Transcriptomic Analysis in Different Parts of Paphiopedilum purpuratum[J]. Biotechnology Bulletin, 2026, 42(6): 279-293.
| 类型 Type | SL vs. SF | SL vs. SR | SF vs. SR | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 上调 Up | 下调 Down | 上调 Up | 下调 Down | 上调 Up | 下调 Down | |||||||
| Positive | Negative | Positive | Negative | Positive | Negative | Positive | Negative | Positive | Negative | Positive | Negative | |
| 脂质 Lipids | 86 | 27 | 21 | 18 | 87 | 27 | 20 | 47 | 30 | 14 | 36 | 37 |
| 苯丙类 Phenylpropanoids | 24 | 14 | 23 | 17 | 26 | 10 | 38 | 20 | 27 | 14 | 38 | 17 |
| 有机氧 Organic oxygen compounds | 23 | 7 | 11 | 18 | 15 | 7 | 10 | 14 | 5 | 15 | 6 | 10 |
| 有机酸 Organic acids | 19 | 1 | 27 | 11 | 10 | 4 | 48 | 16 | 13 | 6 | 28 | 7 |
| 有机杂环 Heterocyclic compounds | 25 | 2 | 15 | 9 | 19 | 2 | 25 | 5 | 11 | 3 | 31 | 2 |
| 核苷类 Nucleosides | 5 | 1 | 6 | 8 | 1 | 2 | 8 | 7 | 3 | 5 | 8 | 0 |
| 苯类 Benzenoids | 27 | 2 | 14 | 6 | 21 | 2 | 18 | 11 | 9 | 2 | 22 | 9 |
| 有机氮 Organic nitrogen compounds | 1 | 1 | 10 | 1 | 0 | 1 | 5 | 1 | 4 | 0 | 3 | 0 |
| 生物碱 Alkaloids | 6 | 1 | 1 | 0 | 4 | 2 | 2 | 1 | 2 | 1 | 3 | 0 |
| 木脂素 Lignans | 2 | 1 | 1 | 0 | 1 | 0 | 1 | 4 | 0 | 0 | 3 | 4 |
| 其他类 Others | 101 | 21 | 51 | 27 | 81 | 35 | 94 | 25 | 49 | 25 | 102 | 16 |
| 总计 Total | 319 | 78 | 180 | 115 | 265 | 92 | 269 | 151 | 153 | 85 | 280 | 102 |
Table 1 Differential expressions of metabolites in different tissues
| 类型 Type | SL vs. SF | SL vs. SR | SF vs. SR | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 上调 Up | 下调 Down | 上调 Up | 下调 Down | 上调 Up | 下调 Down | |||||||
| Positive | Negative | Positive | Negative | Positive | Negative | Positive | Negative | Positive | Negative | Positive | Negative | |
| 脂质 Lipids | 86 | 27 | 21 | 18 | 87 | 27 | 20 | 47 | 30 | 14 | 36 | 37 |
| 苯丙类 Phenylpropanoids | 24 | 14 | 23 | 17 | 26 | 10 | 38 | 20 | 27 | 14 | 38 | 17 |
| 有机氧 Organic oxygen compounds | 23 | 7 | 11 | 18 | 15 | 7 | 10 | 14 | 5 | 15 | 6 | 10 |
| 有机酸 Organic acids | 19 | 1 | 27 | 11 | 10 | 4 | 48 | 16 | 13 | 6 | 28 | 7 |
| 有机杂环 Heterocyclic compounds | 25 | 2 | 15 | 9 | 19 | 2 | 25 | 5 | 11 | 3 | 31 | 2 |
| 核苷类 Nucleosides | 5 | 1 | 6 | 8 | 1 | 2 | 8 | 7 | 3 | 5 | 8 | 0 |
| 苯类 Benzenoids | 27 | 2 | 14 | 6 | 21 | 2 | 18 | 11 | 9 | 2 | 22 | 9 |
| 有机氮 Organic nitrogen compounds | 1 | 1 | 10 | 1 | 0 | 1 | 5 | 1 | 4 | 0 | 3 | 0 |
| 生物碱 Alkaloids | 6 | 1 | 1 | 0 | 4 | 2 | 2 | 1 | 2 | 1 | 3 | 0 |
| 木脂素 Lignans | 2 | 1 | 1 | 0 | 1 | 0 | 1 | 4 | 0 | 0 | 3 | 4 |
| 其他类 Others | 101 | 21 | 51 | 27 | 81 | 35 | 94 | 25 | 49 | 25 | 102 | 16 |
| 总计 Total | 319 | 78 | 180 | 115 | 265 | 92 | 269 | 151 | 153 | 85 | 280 | 102 |
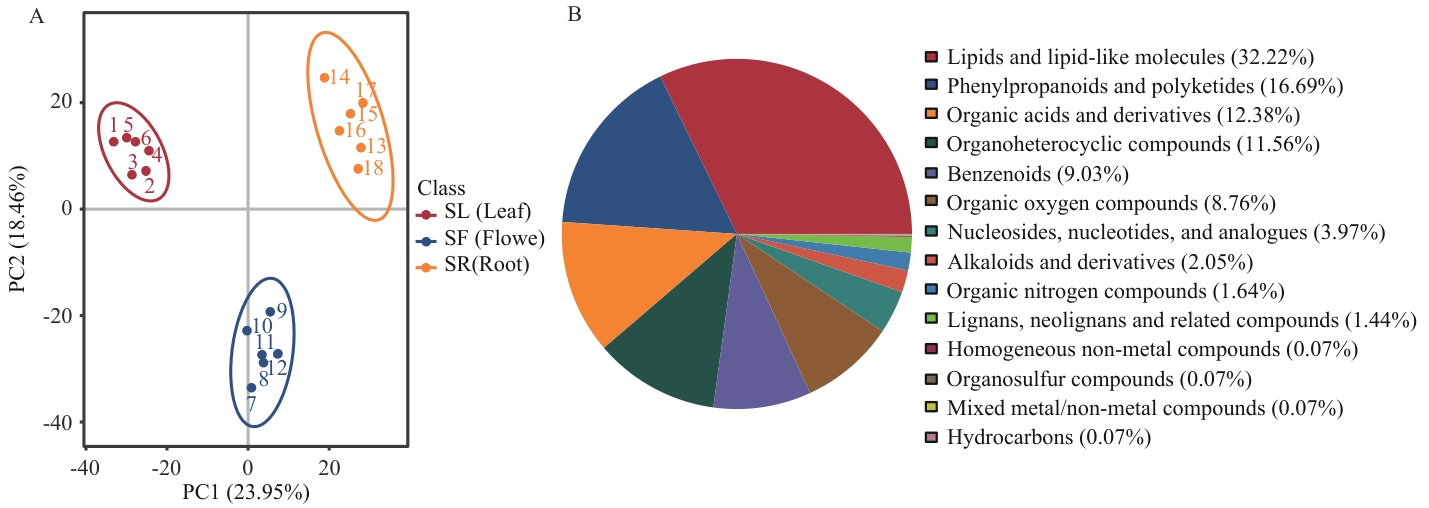
Fig. 1 Classification of metabolites of P. purpuratumA: PCA analysis diagram of the total sample. B: Pie chart for metabolite classification. SL: Leaf. SF: Flower. SR: Root. The same below
代谢物类别 Metabolite category | 代谢物名称 Metabolite name | SL vs. SF | SL vs. SR | SF vs. SR | |||
|---|---|---|---|---|---|---|---|
| Log2FC | VIP | Log2FC | VIP | Log2FC | VIP | ||
| 脂质和类脂分子 Lipids and lipid-like molecules | 姜糖脂B Gingerglycolipid B | 1.95 | 1.52 | 6.92 | 1.19 | 4.96 | 1.07 |
| 重楼皂苷 VI Polyphyllin VI | 2.51 | 1.36 | 6.36 | 1.63 | 3.85 | 1.34 | |
| 柴胡皂苷C Saikosaponin C | 5.72 | 1.66 | 6.06 | 1.05 | / | / | |
| 单酰甘油(18∶3) MAG (18∶3) | / | / | 6.01 | 1.31 | 5.17 | 1.71 | |
| 海葱苷A Proscillaridin A | 5.07 | 1.68 | 6.01 | 1.47 | / | / | |
| 知母皂苷A3 Timosaponin A3 | / | / | 5.95 | 1.39 | 5.26 | 1.50 | |
| 脱氧皮质酮21-葡萄糖苷 Deoxycorticosterone 21-glucoside | 2.52 | 1.76 | 5.76 | 1.86 | 3.23 | 1.25 | |
| 羟基积雪草苷 Madecassoside | 5.31 | 1.41 | 2.78 | 1.50 | / | / | |
| 远志酸 Polygalic acid | 3.18 | 1.60 | 5.20 | 1.79 | / | / | |
| 香叶基丙酮 Geranylacetone | 4.55 | 1.66 | 5.13 | 1.07 | / | / | |
| 二十二碳六烯酸 Docosahexaenoic acid | 4.93 | 1.00 | 3.80 | 1.17 | / | / | |
| 20-羟基蜕皮酮 20-Hydroxyecdysone | 4.05 | 1.43 | 4.88 | 1.14 | / | / | |
| 二氢玫瑰苷 Dihydroroseoside | 4.58 | 1.84 | 4.67 | 1.12 | / | / | |
| 茯苓酸B Poricoic acid B | / | / | 4.67 | 1.01 | 3.84 | 1.52/ | |
| 草乌甲素A Bulleyaconi cine A | / | / | -3.66 | 1.18 | -4.85 | 2.10 | |
| 拉帕醇 Lapachol | -2.75 | 1.07 | -3.81 | 2.68 | / | / | |
| 丹参酚C Danshenol C | / | / | -4.06 | 1.54 | -4.14 | 1.74 | |
| 桦木酮酸 Betulonic acid | 2.34 | 1.17 | / | / | -4.32 | 2.24 | |
| 乙酰哈帕苷 Acetylharpagide | / | / | -3.59 | 1.47 | -4.41 | 1.06 | |
| 毒毛旋花苷 Ouabain | / | / | -4.54 | 1.38 | -4.34 | 1.66 | |
| 竹节参皂苷 IVa Chikusetsusponin IVa | 3.69 | 1.67 | -1.98 | 1.69 | -4.80 | 1.10 | |
| 8-异前列腺素A2 8-iso Prostaglandin A2 | / | / | -4.86 | 1.73 | -5.02 | 1.72 | |
| 马鞭草苷 Verbenalin | 1.16 | 1.04 | -5.09 | 1.15 | -6.25 | 2.02 | |
| 亚油酸 Linoleic acid | / | / | -4.58 | 1.53 | -5.61 | 2.07 | |
| 玫瑰苷 Roseoside | / | / | 4.67 | 1.12 | -6.34 | 1.30 | |
| (±)-脱落酸 (±)-Abscisic acid | / | / | -6.28 | 1.49 | -6.46 | 1.47 | |
| 苯丙类和聚酮类 Phenylpropanoids and polyketides | 槲皮素-3β-D-葡萄糖苷 Quercetin-3β-D-glucoside | -7.65 | 1.84 | / | / | 8.26 | 1.31 |
| 山奈酚 Kaempferol | / | / | 7.27 | 1.63 | 6.60 | 1.29 | |
| 荭草素 Orientin | -1.97 | 1.13 | 3.84 | 1.13 | 5.81 | 1.84 | |
| 矢车菊素-3-O-葡萄糖苷 Kuromanin | / | / | 5.24 | 1.33 | 5.15 | 1.74 | |
| 柚皮素 Naringenin | -3.70 | 1.45 | / | / | 2.65 | 1.06 | |
| 咖啡酸乙酯 Ethyl caffeate | / | / | -4.13 | 1.93 | -3.10 | 1.47 | |
| 落新妇苷 Astilbin | -1.65 | 1.30 | -4.57 | 1.48 | / | / | |
| 异茴芹素 Isopimpinellin | -4.71 | 2.03 | / | / | 4.46 | 1.00 | |
| 曲克芦丁 Troxerutin | -5.37 | 1.48 | -5.72 | 1.61 | / | / | |
| 车前苷 Plantagoside | -6.19 | 1.28 | / | / | 3.64 | 1.32 | |
| 毛蕊花糖苷 Verbascoside | / | / | -6.41 | 1.32 | -4.20 | 1.54 | |
| 槲皮素3-O-槐糖苷 Quercetin 3-O-sophoroside | -9.93 | 1.20 | / | / | 9.64 | 1.91 | |
Table 2 Information on the main significant differential metabolites in different parts of the P. purpuratum
代谢物类别 Metabolite category | 代谢物名称 Metabolite name | SL vs. SF | SL vs. SR | SF vs. SR | |||
|---|---|---|---|---|---|---|---|
| Log2FC | VIP | Log2FC | VIP | Log2FC | VIP | ||
| 脂质和类脂分子 Lipids and lipid-like molecules | 姜糖脂B Gingerglycolipid B | 1.95 | 1.52 | 6.92 | 1.19 | 4.96 | 1.07 |
| 重楼皂苷 VI Polyphyllin VI | 2.51 | 1.36 | 6.36 | 1.63 | 3.85 | 1.34 | |
| 柴胡皂苷C Saikosaponin C | 5.72 | 1.66 | 6.06 | 1.05 | / | / | |
| 单酰甘油(18∶3) MAG (18∶3) | / | / | 6.01 | 1.31 | 5.17 | 1.71 | |
| 海葱苷A Proscillaridin A | 5.07 | 1.68 | 6.01 | 1.47 | / | / | |
| 知母皂苷A3 Timosaponin A3 | / | / | 5.95 | 1.39 | 5.26 | 1.50 | |
| 脱氧皮质酮21-葡萄糖苷 Deoxycorticosterone 21-glucoside | 2.52 | 1.76 | 5.76 | 1.86 | 3.23 | 1.25 | |
| 羟基积雪草苷 Madecassoside | 5.31 | 1.41 | 2.78 | 1.50 | / | / | |
| 远志酸 Polygalic acid | 3.18 | 1.60 | 5.20 | 1.79 | / | / | |
| 香叶基丙酮 Geranylacetone | 4.55 | 1.66 | 5.13 | 1.07 | / | / | |
| 二十二碳六烯酸 Docosahexaenoic acid | 4.93 | 1.00 | 3.80 | 1.17 | / | / | |
| 20-羟基蜕皮酮 20-Hydroxyecdysone | 4.05 | 1.43 | 4.88 | 1.14 | / | / | |
| 二氢玫瑰苷 Dihydroroseoside | 4.58 | 1.84 | 4.67 | 1.12 | / | / | |
| 茯苓酸B Poricoic acid B | / | / | 4.67 | 1.01 | 3.84 | 1.52/ | |
| 草乌甲素A Bulleyaconi cine A | / | / | -3.66 | 1.18 | -4.85 | 2.10 | |
| 拉帕醇 Lapachol | -2.75 | 1.07 | -3.81 | 2.68 | / | / | |
| 丹参酚C Danshenol C | / | / | -4.06 | 1.54 | -4.14 | 1.74 | |
| 桦木酮酸 Betulonic acid | 2.34 | 1.17 | / | / | -4.32 | 2.24 | |
| 乙酰哈帕苷 Acetylharpagide | / | / | -3.59 | 1.47 | -4.41 | 1.06 | |
| 毒毛旋花苷 Ouabain | / | / | -4.54 | 1.38 | -4.34 | 1.66 | |
| 竹节参皂苷 IVa Chikusetsusponin IVa | 3.69 | 1.67 | -1.98 | 1.69 | -4.80 | 1.10 | |
| 8-异前列腺素A2 8-iso Prostaglandin A2 | / | / | -4.86 | 1.73 | -5.02 | 1.72 | |
| 马鞭草苷 Verbenalin | 1.16 | 1.04 | -5.09 | 1.15 | -6.25 | 2.02 | |
| 亚油酸 Linoleic acid | / | / | -4.58 | 1.53 | -5.61 | 2.07 | |
| 玫瑰苷 Roseoside | / | / | 4.67 | 1.12 | -6.34 | 1.30 | |
| (±)-脱落酸 (±)-Abscisic acid | / | / | -6.28 | 1.49 | -6.46 | 1.47 | |
| 苯丙类和聚酮类 Phenylpropanoids and polyketides | 槲皮素-3β-D-葡萄糖苷 Quercetin-3β-D-glucoside | -7.65 | 1.84 | / | / | 8.26 | 1.31 |
| 山奈酚 Kaempferol | / | / | 7.27 | 1.63 | 6.60 | 1.29 | |
| 荭草素 Orientin | -1.97 | 1.13 | 3.84 | 1.13 | 5.81 | 1.84 | |
| 矢车菊素-3-O-葡萄糖苷 Kuromanin | / | / | 5.24 | 1.33 | 5.15 | 1.74 | |
| 柚皮素 Naringenin | -3.70 | 1.45 | / | / | 2.65 | 1.06 | |
| 咖啡酸乙酯 Ethyl caffeate | / | / | -4.13 | 1.93 | -3.10 | 1.47 | |
| 落新妇苷 Astilbin | -1.65 | 1.30 | -4.57 | 1.48 | / | / | |
| 异茴芹素 Isopimpinellin | -4.71 | 2.03 | / | / | 4.46 | 1.00 | |
| 曲克芦丁 Troxerutin | -5.37 | 1.48 | -5.72 | 1.61 | / | / | |
| 车前苷 Plantagoside | -6.19 | 1.28 | / | / | 3.64 | 1.32 | |
| 毛蕊花糖苷 Verbascoside | / | / | -6.41 | 1.32 | -4.20 | 1.54 | |
| 槲皮素3-O-槐糖苷 Quercetin 3-O-sophoroside | -9.93 | 1.20 | / | / | 9.64 | 1.91 | |
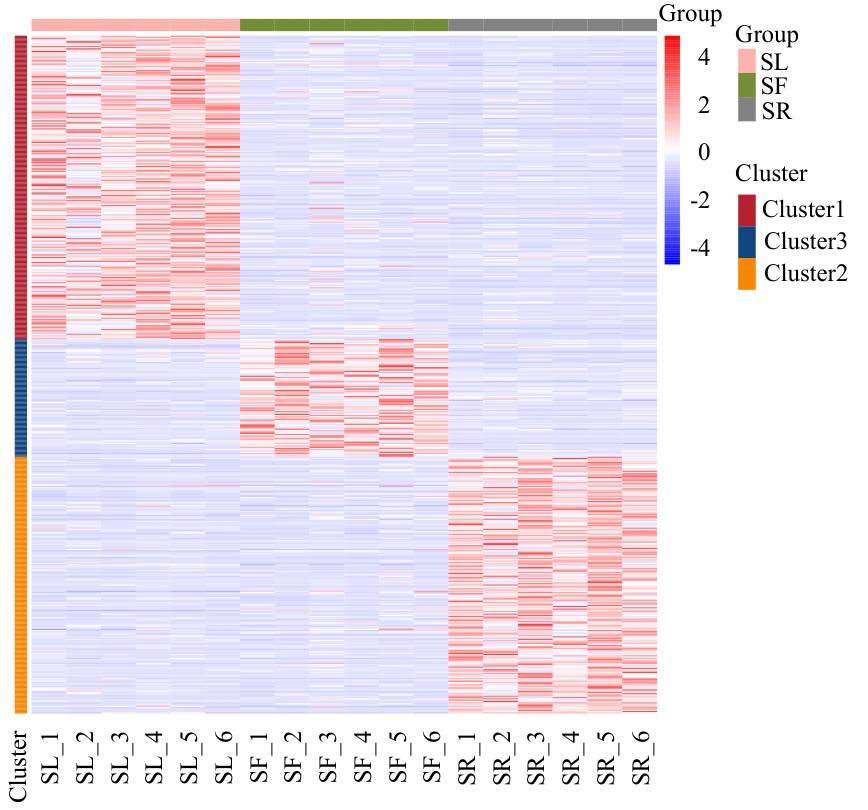
Fig. 2 Clustering heatmap of differential metabolitesRed indicates up-regulation, and blue indicates down-regulation. The same below
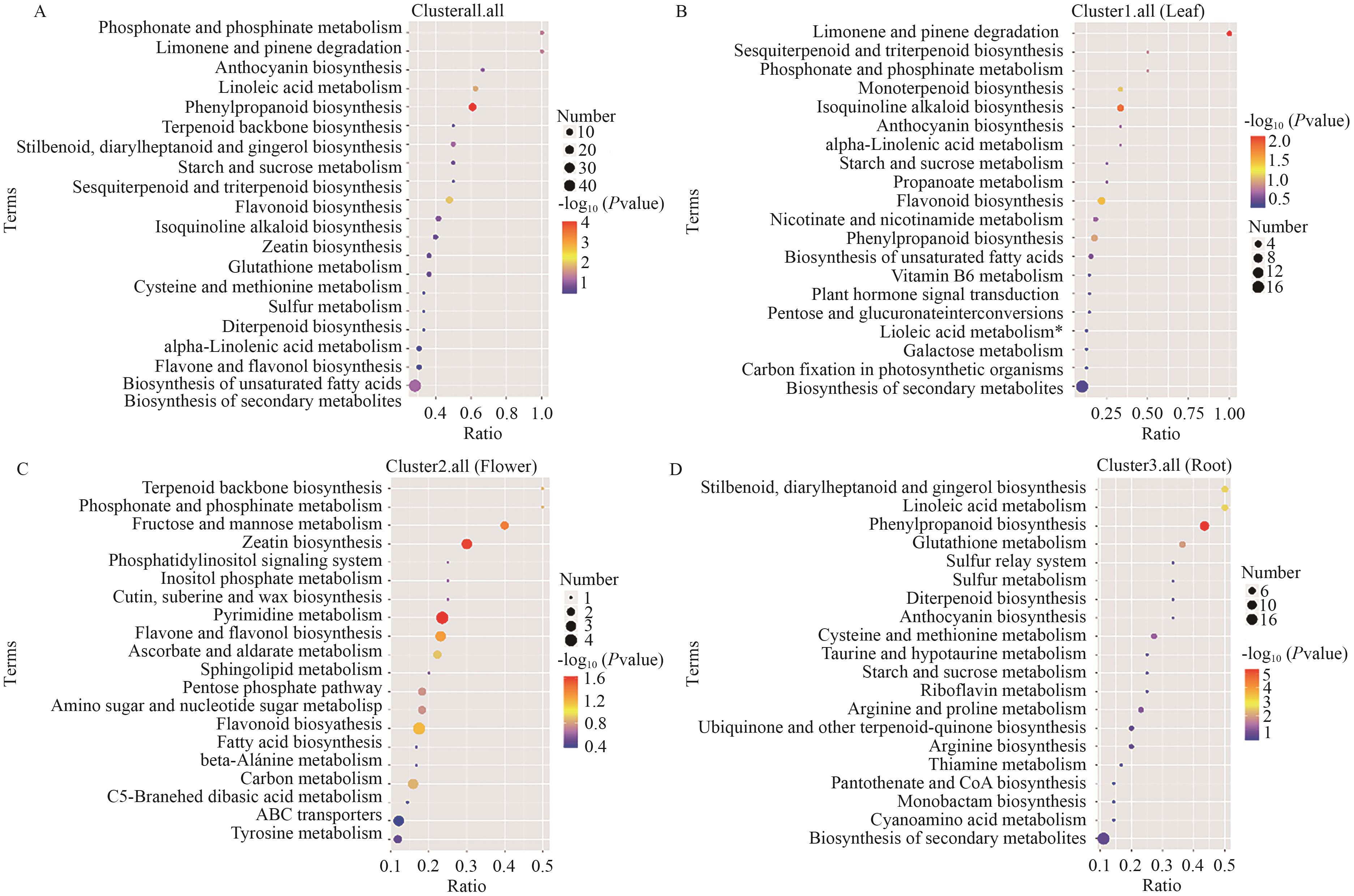
Fig. 3 Non-targeted KEGG enrichment bubble chartBiosynthetic pathways significantly enriched with differential metabolites. A: All parts. B: Leaves. C: Flowers. D: Roots
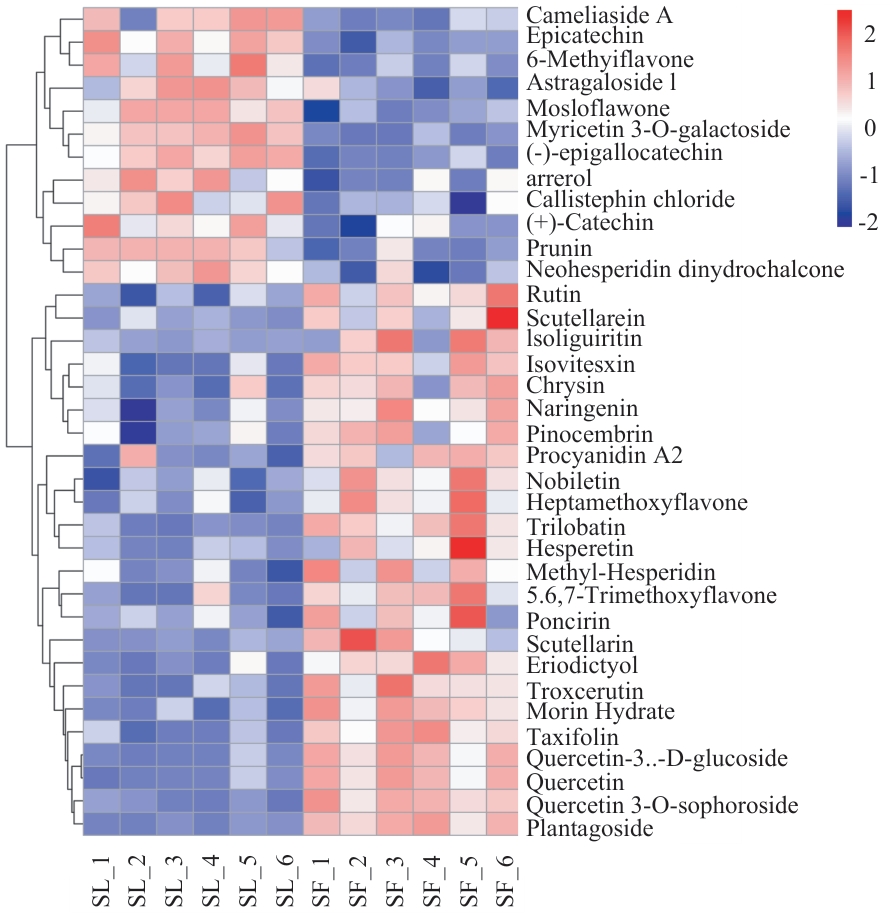
Fig. 4 Heatmap of flavonoid metabolites in flowers and leaves
Fig. 5 Consensus statistical analysis chart
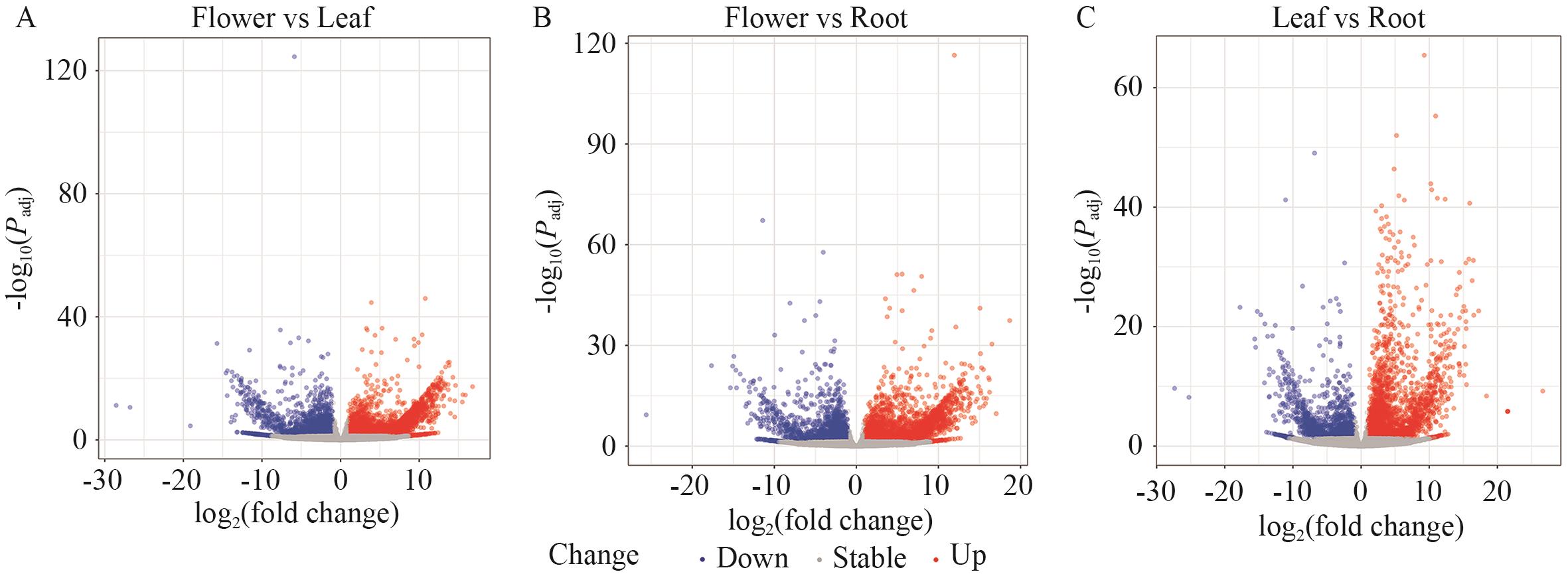
Fig. 6 Volcano plot of differentially expressed genes (DEGs) in different parts of P. purpuratum
| 比较组 Comparison group | 调控方向 Up/Down | 基因Gene | Log2FC | FDR | 功能注释 Functional annotation |
|---|---|---|---|---|---|
| SL vs. SF | 上调 Up | GLDC | 15.72 | 4.40e-32 | 碳代谢 Carbon metabolism |
| 上调 Up | LOX1_5 | 14.59 | 1.74e-22 | 脂质代谢和亚油酸代谢 Lipid metabolism and linoleic acid metabolism | |
| 上调 Up | chlH | 14.42 | 4.33e-15 | 叶绿素合成 Chlorophyll synthesis | |
| 上调 Up | cynT | 13.94 | 1.61e-6 | 氮代谢 Nitrogen metabolism | |
| 上调 Up | PMA1/PMA2 | 13.51 | 4.38e-7 | 能量代谢 Energy metabolism | |
| 下调 Down | GPAT | -10.00 | 1.14e-10 | 脂质代谢 Lipid metabolism | |
| 下调 Down | P5CS | -9.98 | 1.37e-11 | 氨基酸代谢 Amino acid metabolism | |
| 下调 Down | pdhC | -9.95 | 1.23e-7 | 碳代谢 Carbon metabolism | |
| 下调 Down | ALDH | -9.92 | 2.47e-9 | 新陈代谢 Metabolism | |
| 下调 Down | PIP5K | -9.85 | 5.33e-12 | 信号转导 Signal transduction | |
| SL vs. SR | 上调 Up | nadB | 9.99 | 3.65e-12 | 新陈代谢 Metabolism |
| 上调 Up | ALDO | 9.30 | 1.34e-07 | 新陈代谢 Metabolism | |
| 下调 Down | E1.11.1.7 | -14.72 | 1.04e-22 | 次生代谢物的生物合成 Biosynthesis of secondary metabolites | |
| 下调 Down | CISZOG | -14.18 | 2.63e-11 | 萜类和多酮类化合物的代谢 Terpenoid and polyketide metabolism | |
| 下调 Down | PAL | -13.46 | 4.69e-9 | 新陈代谢 Metabolism | |
| 下调 Down | TGA | -11.80 | 1.37e-7 | 植物激素信号转导 Plant hormone signal transduction | |
| SF vs. SR | 上调 Up | psbS | 10.53 | 1.04e-05 | 新陈代谢 Metabolism |
| 上调 Up | FAH | 8.10 | 1.61e-06 | 脂质代谢 Lipid metabolism | |
| 上调 Up | crtZ | 6.94 | 3.17e-03 | 次生代谢物的生物合成 Biosynthesis of secondary metabolites | |
| 下调 Down | ROMT | -15.07 | 1.49e-24 | 次生代谢物的生物合成 Biosynthesis of secondary metabolites | |
| 下调 Down | HHT1 | -14.69 | 2.42e-23 | 脂质代谢 Lipid metabolism | |
| 下调 Down | asnB | -13.24 | 1.58e-07 | 氨基酸代谢 Amino acid metabolism | |
| 下调 Down | SGT1 | -12.89 | 1.74e-08 | 聚糖的生物合成与代谢 Glycan biosynthesis and metabolism | |
| 下调 Down | LOX1_5 | -8.82 | 5.84e-09 | 脂质代谢和亚油酸代谢 Lipid metabolism and linoleic acid metabolism |
Table 3 Main significant differential gene information in different parts of P. purpuratum
| 比较组 Comparison group | 调控方向 Up/Down | 基因Gene | Log2FC | FDR | 功能注释 Functional annotation |
|---|---|---|---|---|---|
| SL vs. SF | 上调 Up | GLDC | 15.72 | 4.40e-32 | 碳代谢 Carbon metabolism |
| 上调 Up | LOX1_5 | 14.59 | 1.74e-22 | 脂质代谢和亚油酸代谢 Lipid metabolism and linoleic acid metabolism | |
| 上调 Up | chlH | 14.42 | 4.33e-15 | 叶绿素合成 Chlorophyll synthesis | |
| 上调 Up | cynT | 13.94 | 1.61e-6 | 氮代谢 Nitrogen metabolism | |
| 上调 Up | PMA1/PMA2 | 13.51 | 4.38e-7 | 能量代谢 Energy metabolism | |
| 下调 Down | GPAT | -10.00 | 1.14e-10 | 脂质代谢 Lipid metabolism | |
| 下调 Down | P5CS | -9.98 | 1.37e-11 | 氨基酸代谢 Amino acid metabolism | |
| 下调 Down | pdhC | -9.95 | 1.23e-7 | 碳代谢 Carbon metabolism | |
| 下调 Down | ALDH | -9.92 | 2.47e-9 | 新陈代谢 Metabolism | |
| 下调 Down | PIP5K | -9.85 | 5.33e-12 | 信号转导 Signal transduction | |
| SL vs. SR | 上调 Up | nadB | 9.99 | 3.65e-12 | 新陈代谢 Metabolism |
| 上调 Up | ALDO | 9.30 | 1.34e-07 | 新陈代谢 Metabolism | |
| 下调 Down | E1.11.1.7 | -14.72 | 1.04e-22 | 次生代谢物的生物合成 Biosynthesis of secondary metabolites | |
| 下调 Down | CISZOG | -14.18 | 2.63e-11 | 萜类和多酮类化合物的代谢 Terpenoid and polyketide metabolism | |
| 下调 Down | PAL | -13.46 | 4.69e-9 | 新陈代谢 Metabolism | |
| 下调 Down | TGA | -11.80 | 1.37e-7 | 植物激素信号转导 Plant hormone signal transduction | |
| SF vs. SR | 上调 Up | psbS | 10.53 | 1.04e-05 | 新陈代谢 Metabolism |
| 上调 Up | FAH | 8.10 | 1.61e-06 | 脂质代谢 Lipid metabolism | |
| 上调 Up | crtZ | 6.94 | 3.17e-03 | 次生代谢物的生物合成 Biosynthesis of secondary metabolites | |
| 下调 Down | ROMT | -15.07 | 1.49e-24 | 次生代谢物的生物合成 Biosynthesis of secondary metabolites | |
| 下调 Down | HHT1 | -14.69 | 2.42e-23 | 脂质代谢 Lipid metabolism | |
| 下调 Down | asnB | -13.24 | 1.58e-07 | 氨基酸代谢 Amino acid metabolism | |
| 下调 Down | SGT1 | -12.89 | 1.74e-08 | 聚糖的生物合成与代谢 Glycan biosynthesis and metabolism | |
| 下调 Down | LOX1_5 | -8.82 | 5.84e-09 | 脂质代谢和亚油酸代谢 Lipid metabolism and linoleic acid metabolism |
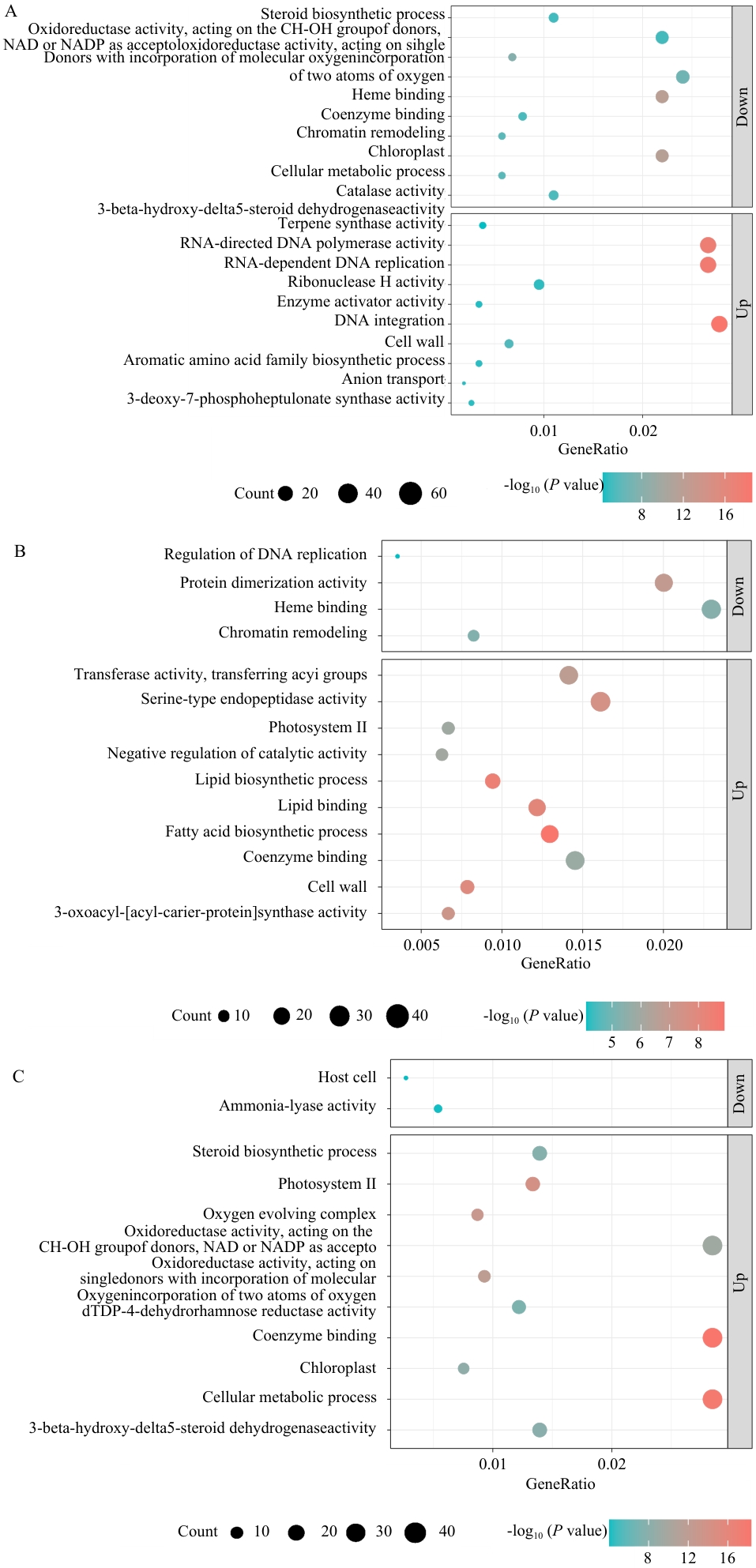
Fig. 7 GO functional enrichment analysis of DEGs in different parts of P. purpuratum
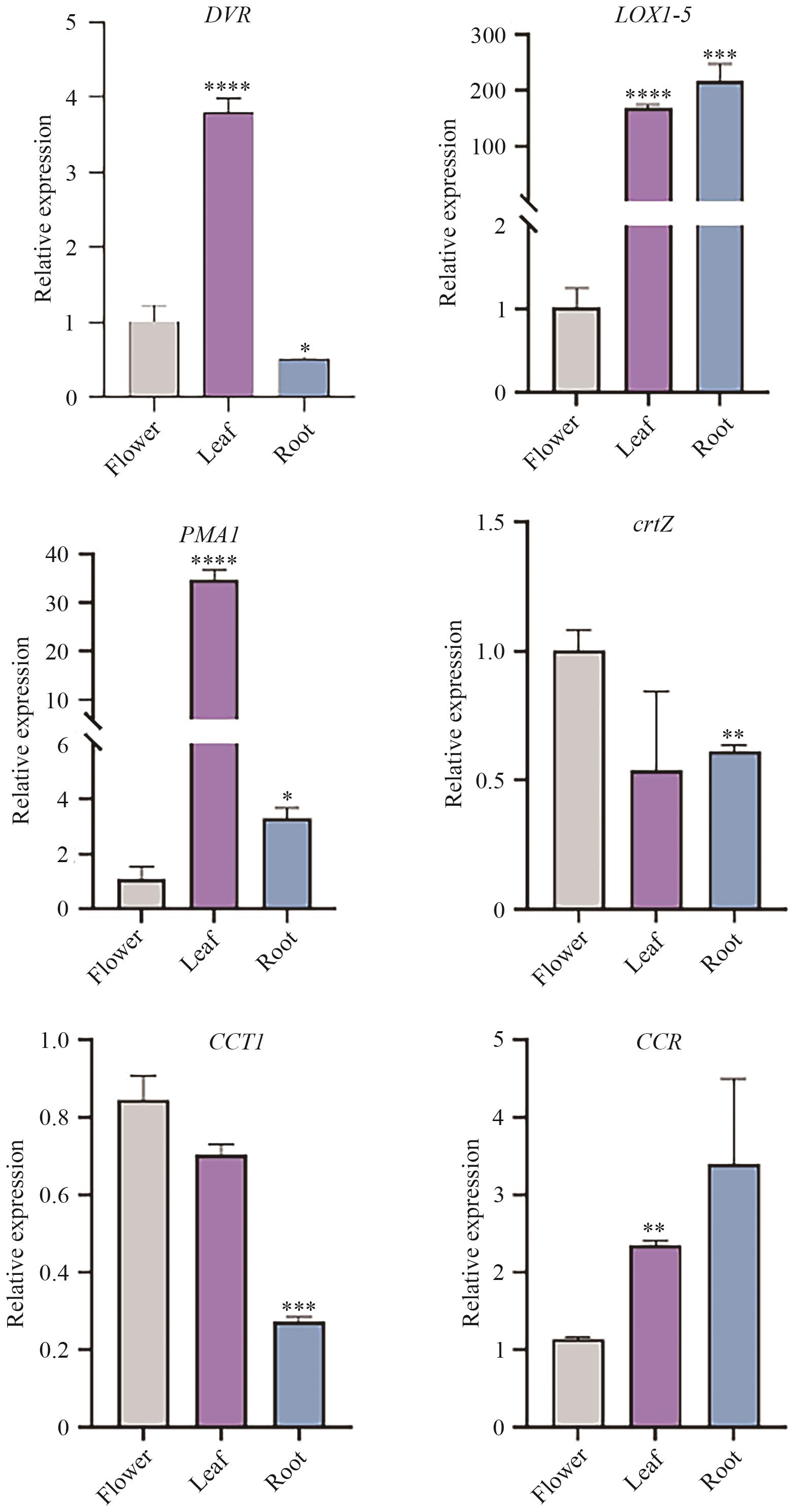
Fig. 8 RT-qPCR analysis of six DEGs
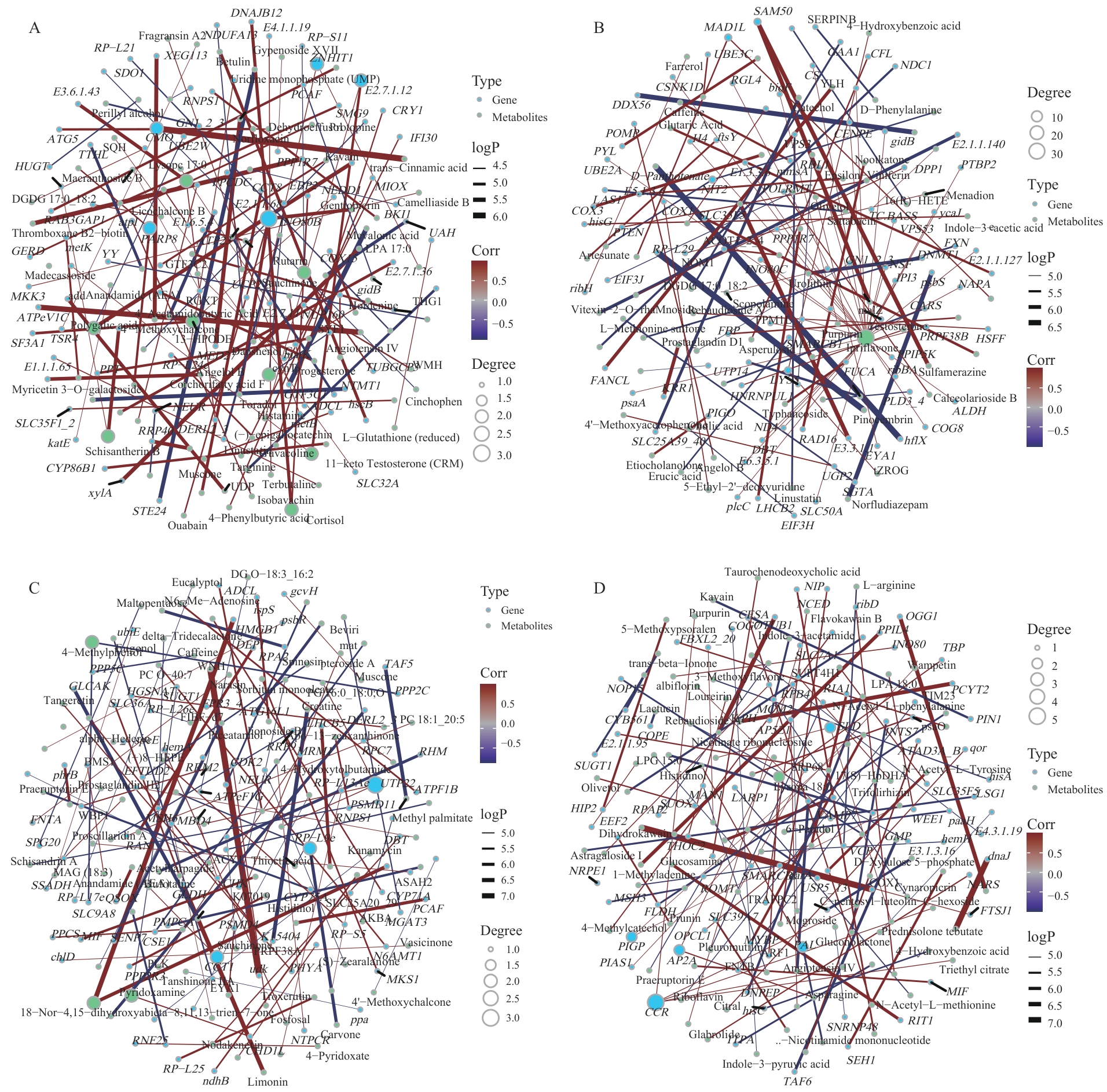
Fig. 9 Correlation analysis between metabolome and transcriptome in different parts of P. purpuratumA: All parts. B: Leaves. C: Flowers. D: Roots
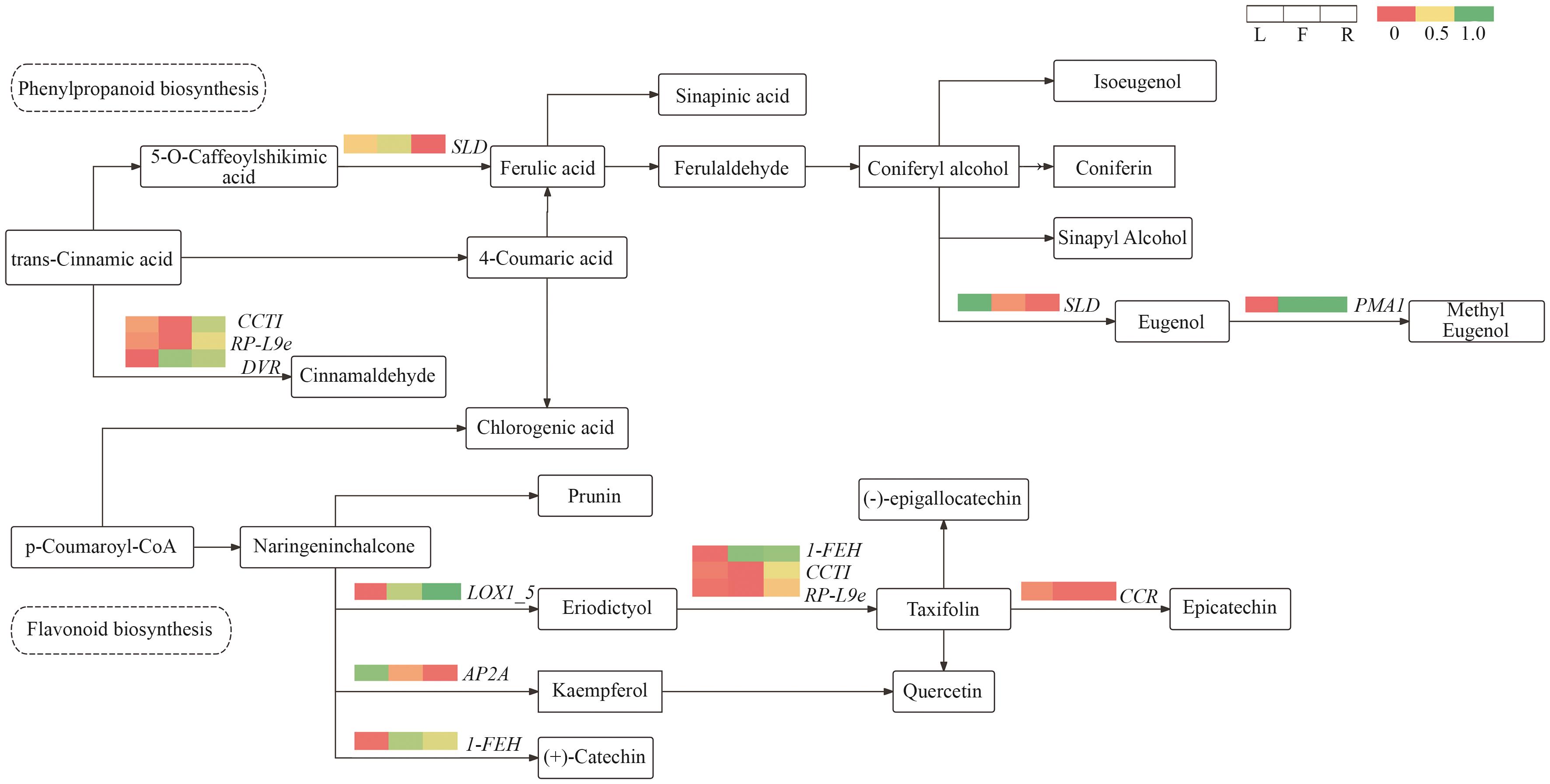
Fig. 10 Changes in metabolites and genes at hubs of the co-expression networkThe heatmap indicates the expressions of differentially expressed genes across different tissues. Red color indicates high expression level, and green color indicates low expression level
| [1] | 罗毅波, 贾建生, 王春玲. 初论中国兜兰属植物的保护策略及其潜在资源优势 [J]. 生物多样性, 2003, 11(6): 491-498. |
| Luo YB, Jia JS, Wang CL. Conservation strategy and potential advantages of the Chinese Paphiopedilum [J]. Chin Biodivers, 2003, 11(6): 491-498. | |
| [2] | 刘仲健, 陈心启, 陈利君, 等. 中国兜兰属植物 [M]. 北京: 科学出版社, 2009. |
| Liu ZJ, Chen XQ, Chen LJ, et al. The genus Paphiopedilum in China [M]. Beijing: Science Press, 2009. | |
| [3] | Sun JJ, Li QL, Xu H, et al. Analysis of metabolomic changes in xylem and phloem sap of cucumber under phosphorus stresses [J]. Metabolites, 2022, 12(4): 361. |
| [4] | Zhou ZW, Luo MD, Chen X, et al. Ion mobility collision cross-section atlas for known and unknown metabolite annotation in untargeted metabolomics [J]. Nat Commun, 2020, 11(1): 4334. |
| [5] | Guo AH, Yang Y, Wu J, et al. Lipidomic and transcriptomic profiles of glycerophospholipid metabolism during Hemerocallis citrina Baroni flowering [J]. BMC Plant Biol, 2023, 23(1): 50. |
| [6] | Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics [J]. Nat Rev Genet, 2009, 10(1): 57-63. |
| [7] | Zhao RX, Yan S, Hu YD, et al. Metabolic and transcriptomic profile revealing the differential accumulating mechanism in different parts of Dendrobium nobile [J]. Int J Mol Sci, 2024, 25(10): 5356. |
| [8] | Qiu YJ, Cai CC, Mo X, et al. Transcriptome and metabolome analysis reveals the effect of flavonoids on flower color variation in Dendrobium nobile Lindl [J]. Front Plant Sci, 2023, 14: 1220507. |
| [9] | Ahmad S, Lu CQ, Gao J, et al. Integrated proteomic, transcriptomic, and metabolomic profiling reveals that the gibberellin-abscisic acid hub runs flower development in the Chinese orchid Cymbidium sinense [J]. Hortic Res, 2024, 11(5): uhae073. |
| [10] | Zhang GJ, Hu Y, Huang MZ, et al. Comprehensive phylogenetic analyses of Orchidaceae using nuclear genes and evolutionary insights into epiphytism [J]. J Integr Plant Biol, 2023, 65(5): 1204-1225. |
| [11] | 王蒙, 王婷, 夏增强, 等. 基于转录组数据揭示4种兜兰的全基因组复制历史 [J]. 植物学报, 2021, 56(6): 699-714. |
| Wang M, Wang T, Xia ZQ, et al. Revealing the new whole-genome duplication event of four Paphiopedilum species based on transcriptome data [J]. Chin Bull Bot, 2021, 56(6): 699-714. | |
| [12] | Liang YY, Hao J, Wang JY, et al. Statistical genomics analysis of simple sequence repeats from the Paphiopedilum malipoense transcriptome reveals control knob motifs modulating gene expression [J]. Adv Sci, 2024, 11(24): 2304848. |
| [13] | Ye YQ, Chang YT, Ma YJ, et al. Comparative study on the mechanism of yellow petal formation in Paphiopedilum armeniacum: an integrated transcriptomic and metabolomic analysis of three Paphiopedilum species [J]. BMC Genomics, 2025, 26(1): 560. |
| [14] | Wen B, Mei ZL, Zeng CW, et al. metaX: a flexible and comprehensive software for processing metabolomics data [J]. BMC Bioinformatics, 2017, 18(1): 183. |
| [15] | Haspel JA, Chettimada S, Shaik RS, et al. Circadian rhythm reprogramming during lung inflammation [J]. Nat Commun, 2014, 5: 4753. |
| [16] | Sreekumar A, Poisson LM, Rajendiran TM, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression [J]. Nature, 2009, 457(7231): 910-914. |
| [17] | Chen SF, Zhou YQ, Chen YR, et al. Fastp: an ultra-fast all-in-one FASTQ preprocessor [J]. Bioinformatics, 2018, 34(17): i884-i890. |
| [18] | Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data [J]. Bioinformatics, 2014, 30(15): 2114-2120. |
| [19] | Patro R, Duggal G, Love MI, et al. Salmon provides fast and bias-aware quantification of transcript expression [J]. Nat Meth, 2017, 14(4): 417-419. |
| [20] | Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2 [J]. Genome Biol, 2014, 15(12): 550. |
| [21] | Yu GC, Wang LG, Han YY, et al. clusterProfiler: an R package for comparing biological themes among gene clusters [J]. OMICS, 2012, 16(5): 284-287. |
| [22] | Fan JW, Shen YT, Chen C, et al. A large-scale integrated transcriptomic atlas for soybean organ development [J]. Mol Plant, 2025, 18(4): 669-689. |
| [23] | 孙晓琛, 栗锦鹏, 原静静, 等. 基于转录组测序分析干旱胁迫对党参不同组织基因表达的调控 [J]. 中草药, 2022, 53(14): 4465-4475. |
| Sun XC, Li JP, Yuan JJ, et al. Sequencing and analysis of transcriptome to reveal regulation gene expression in different tissues of Codonopsis pilosula under drought stress [J]. Chin Tradit Herb Drugs, 2022, 53(14): 4465-4475. | |
| [24] | Zhao XC, Wei JP, He L, et al. Identification of fatty acid desaturases in maize and their differential responses to low and high temperature [J]. Genes, 2019, 10(6): 445. |
| [25] | Qiao Q, Wu C, Cheng TT, et al. Comparative analysis of the metabolome and transcriptome between the green and yellow-green regions of variegated leaves in a mutant variety of the tree species Pteroceltis tatarinowii [J]. Int J Mol Sci, 2022, 23(9): 4950. |
| [26] | Bao YR, Nie TK, Wang DD, et al. Anthocyanin regulatory networks in Solanum tuberosum L. leaves elucidated via integrated metabolomics, transcriptomics, and StAN1 overexpression [J]. BMC Plant Biol, 2022, 22(1): 228. |
| [27] | Ghasemzadeh A, Jaafar HZE, Rahmat A. Synthesis of phenolics and flavonoids in ginger (Zingiber officinale Roscoe) and their effects on photosynthesis rate [J]. Int J Mol Sci, 2010, 11(11): 4539-4555. |
| [28] | Hong JY, Gunasekara C, He C, et al. Identification of biological pathway and process regulators using sparse partial least squares and triple-gene mutual interaction [J]. Sci Rep, 2021, 11(1): 13174. |
| [29] | He XJ, Zhao XC, Gao LP, et al. Isolation and characterization of key genes that promote flavonoid accumulation in purple-leaf tea (Camellia sinensis L.) [J]. Sci Rep, 2018, 8(1): 130. |
| [30] | Luo JR, Duan JJ, Huo D, et al. Transcriptomic analysis reveals transcription factors related to leaf anthocyanin biosynthesis in Paeonia qiui [J]. Molecules, 2017, 22(12): 2186. |
| [31] | Xiang P, Zhu QF, Tukhvatshin M, et al. Light control of catechin accumulation is mediated by photosynthetic capacity in tea plant (Camellia sinensis) [J]. BMC Plant Biol, 2021, 21(1): 478. |
| [32] | 陈伟, 顿春垚, 李双龙, 等. 不同油茶品种间儿茶素含量差异及油茶炭疽病关系 [J]. 山东农业大学学报: 自然科学版, 2024, 55(5): 733-739. |
| Chen W, Dun CY, Li SL, et al. Differences in catechin content among different Camellia oleifera varieties and connection [J]. J Shandong Agric Univ Nat Sci Ed, 2024, 55(5): 733-739. | |
| [33] | Sohlenkamp C, Geiger O. Bacterial membrane lipids: diversity in structures and pathways [J]. FEMS Microbiol Rev, 2016, 40(1): 133-159. |
| [34] | Du K, Jiang SX, Chen H, et al. Spatiotemporal miRNA and transcriptomic network dynamically regulate the developmental and senescence processes of poplar leaves [J]. Hortic Res, 2023, 10(10): uhad186. |
| [35] | Dun HF, Hung TH, Green S, et al. Comparative transcriptomic responses of European and Japanese larches to infection by Phytophthora ramorum [J]. BMC Plant Biol, 2022, 22(1): 480. |
| [36] | Wang J, Cao K, Wang LR, et al. Two MYB and three bHLH family genes participate in anthocyanin accumulation in the flesh of peach fruit treated with glucose, sucrose, sorbitol, and fructose in vitro [J]. Plants, 2022, 11(4): 507. |
| [37] | Peng YY, Thrimawithana AH, Cooney JM, et al. The proanthocyanin-related transcription factors MYBC1 and WRKY44 regulate branch points in the kiwifruit anthocyanin pathway [J]. Sci Rep, 2020, 10(1): 14161. |
| [38] | Tu MX, Fang JH, Zhao RK, et al. CRISPR/Cas9-mediated mutagenesis of VvbZIP36 promotes anthocyanin accumulation in grapevine (Vitis vinifera) [J]. Hortic Res, 2022, 9: uhac022. |
| [39] | Mbanjo EGN, Kretzschmar T, Jones H, et al. The genetic basis and nutritional benefits of pigmented rice grain [J]. Front Genet, 2020, 11: 229. |
| [40] | Hong H, Seo H, Park W, et al. Sequence, structure and function-based classification of the broadly conserved FAH superfamily reveals two distinct fumarylpyruvate hydrolase subfamilies [J]. Environ Microbiol, 2020, 22(1): 270-285. |
| [41] | Athmika, Ghate SD, Arun AB, et al. Genome analysis of a halophilic bacterium Halomonas malpeensis YU-PRIM-29(T) reveals its exopolysaccharide and pigment producing capabilities [J]. Sci Rep, 2021, 11(1): 1749. |
| [42] | Iwasaka H, Koyanagi R, Satoh R, et al. A possible trifunctional β-carotene synthase gene identified in the draft genome of Aurantiochytrium sp. strain KH105 [J]. Genes, 2018, 9(4): 200. |
| [43] | Chuang YC, Lee MC, Chang YL, et al. Diurnal regulation of the floral scent emission by light and circadian rhythm in the Phalaenopsis orchids [J]. Bot Stud, 2017, 58(1): 50. |
| [44] | Jin JJ, Kim MJ, Dhandapani S, et al. The floral transcriptome of ylang ylang (Cananga odorata var. fruticosa) uncovers biosynthetic pathways for volatile organic compounds and a multifunctional and novel sesquiterpene synthase [J]. J Exp Bot, 2015, 66(13): 3959-3975. |
| [45] | Krug C, Cordeiro GD, Schäffler I, et al. Nocturnal bee pollinators are attracted to guarana flowers by their scents [J]. Front Plant Sci, 2018, 9: 1072. |
| [46] | Wu D, Yu L, Nair MG, et al. Cyclooxygenase enzyme inhibitory compounds with antioxidant activities from Piper methysticum (kava kava) roots [J]. Phytomedicine, 2002, 9(1): 41-47. |
| [47] | Gakière B, Hao JF, de Bont L, et al. NAD+ biosynthesis and signaling in plants [J]. Crit Rev Plant Sci, 2018, 37(4): 259-307. |
| [48] | Zhu FD, Fu X, Ye HC, et al. Antibacterial activities of coumarin-3-carboxylic acid against Acidovorax citrulli [J]. Front Microbiol, 2023, 14: 1207125. |
| [49] | 陈荷莹, 刘毅, 刘会珍, 等. 落新妇苷的稳定性及生物活性研究进展 [J]. 世界中医药, 2023, 18(11): 1609-1614. |
| Chen HY, Liu Y, Liu HZ, et al. Research progress on the stability and biological activity of astilbin [J]. World Chin Med, 2023, 18(11): 1609-1614. | |
| [50] | Cao Y, Du PH, Zhang JR, et al. Dopamine alleviates cadmium stress in apple trees by recruiting beneficial microorganisms to enhance the physiological resilience revealed by high-throughput sequencing and soil metabolomics [J]. Hortic Res, 2023, 10(7): uhad112. |
| [51] | Singh AK, Dhanapal S, Yadav BS. The dynamic responses of plant physiology and metabolism during environmental stress progression [J]. Mol Biol Rep, 2020, 47(2): 1459-1470. |
| [52] | Li WL, Lee J, Yu S, et al. Characterization and analysis of the transcriptome response to drought in Larix kaempferi using PacBio full-length cDNA sequencing integrated with de novo RNA-seq reads [J]. Planta, 2021, 253(2): 28. |
| [53] | Zou S, Lu YC, Ma HY, et al. Microalgal glycerol-3-phosphate acyltransferase role in galactolipids and high-value storage lipid biosynthesis [J]. Plant Physiol, 2023, 192(1): 426-441. |
| [54] | Li R, Zhao Y, Sun Z, et al. Genome-wide identification of switchgrass laccases involved in lignin biosynthesis and heavy-metal responses [J]. Int J Mol Sci, 2022, 23(12): 6530. |
| [55] | Weremczuk-Jeżyna I, Hnatuszko-Konka K, Lebelt L, et al. The protective function and modification of secondary metabolite accumulation in response to light stress in Dracocephalum forrestii shoots [J]. Int J Mol Sci, 2021, 22(15): 7965. |
| [56] | Wang MN, Manzoor MA, Wang XY, et al. Comparative genomic analysis of SAUR gene family, cloning and functional characterization of two genes (PbrSAUR13 and PbrSAUR52) in Pyrus bretschneideri [J]. Int J Mol Sci, 2022, 23(13): 7054. |
| [57] | Zhang LS, Zheng LT, Wu JW, et al. OsCCRL1 is essential for phenylpropanoid metabolism in rice anthers [J]. Rice, 2023, 16(1): 10. |
| [58] | Badri DV, De-la-Peña C, Lei ZT, et al. Root secreted metabolites and proteins are involved in the early events of plant-plant recognition prior to competition [J]. PLoS One, 2012, 7(10): e46640. |
| [59] | Puig J, Pauluzzi G, Guiderdoni E, et al. Regulation of shoot and root development through mutual signaling [J]. Mol Plant, 2012, 5(5): 974-983. |
| [1] | WANG Yu-kun, YUAN Yuan, WANG Bin, ZHU Yun-na, REN Xiao-qiang, REN Fei, YE Hong. Integrated Analysis of Transcriptome and Lipid Metabolome Reveals the Differences in α-Linolenic Acid Synthesis Regulation in Different Perilla frutescens [J]. Biotechnology Bulletin, 2026, 42(4): 129-140. |
| [2] | LIU Qing-yuan, WU Hong-qi, CHEN Xiu-e, CHEN Jian, JIANG Yuan-ze, HE Yan-zi, YU Qi-wei, LIU Ren-xiang. Function of Transcription Factor NtMYB96a in Regulating the Tolerance of Tobacco to Drought [J]. Biotechnology Bulletin, 2026, 42(4): 239-250. |
| [3] | YANG Ting, YANG Zong-tao, AI Jing, WANG Yu-tong, LI Yan-ye, DENG jun, LIU Jia-yong, ZHAO Yong, ZHANG Yue-bin. Analysis of Phenotypic Characteristics and Root Transcriptomics of Sugarcane with Different Genotypes [J]. Biotechnology Bulletin, 2026, 42(4): 190-201. |
| [4] | YIN Yue, QIN Xiao-ya, MI Jia, AN Wei, HE Jun, ZHANG Feng-feng. Identification of FBN Gene Family and Its Relationship with Carotenoids Metabolism in Lyciumbarbarum [J]. Biotechnology Bulletin, 2026, 42(3): 338-348. |
| [5] | WANG Xiao-yi, LI Jin-yan, XING Xing, ZHU Hong-liang. Screening and Functional Analysis of Ethylene-responsive Genes Regulating Tomato Fruit Ripening and Respiration [J]. Biotechnology Bulletin, 2026, 42(3): 275-282. |
| [6] | WANG Shang-feng, CHENG Bin, WANG Ruo-ruo, DING Yan-qing, XU Jian-xia, CAO Ning, GAO Xu, LI Wen-zhen, ZHANG Li-yi. Gene Mapping of Sorghum Flowering Time and Prediction of Candidate Genes Based on BSA-seq [J]. Biotechnology Bulletin, 2026, 42(2): 197-206. |
| [7] | FU Han, SUN Shu-hao, ZHANG Si-qing, AI Niu, YU Yang, YU Lian-wei, WANG Qiong-qiong, HAN Xiao-yu, SHI Yan, HAN Wei-li, YANG Xue. Genome-wide Identification and Expression Analysis of the BOI Gene Family in Nicotiana benthamiana [J]. Biotechnology Bulletin, 2026, 42(2): 207-217. |
| [8] | FAN Rong-hui, LUO Yuan-hua, CHEN Yi-quan, FANG Neng-yan, CHEN Yan, ZHONG Huai-qin, YE Xiu-xian. Comparative Analysis of Floral Scent Formation between ‘Jinhui’ and Onc. ‘Sharry Baby’ of Oncidium hybridum [J]. Biotechnology Bulletin, 2026, 42(1): 105-113. |
| [9] | LIU Jian-guo, LIU Ge-er, GUO Ying-xin, WANG Bin, WANG Yu-kun, LU Jin-feng, HUANG Wen-ting, ZHU Yun-na. Integrate Transcriptomic and Metabolomic Analysis of Fruits Quality Differences between ‘Guiyou No. 1’ and ‘Shatianyou’ Pomelo (Citrus maxima) [J]. Biotechnology Bulletin, 2025, 41(9): 168-181. |
| [10] | LIU Ze-zhou, DUAN Nai-bin, YUE Li-xin, WANG Qing-hua, YAO Xing-hao, GAO Li-min, KONG Su-ping. Analysis of Wax Components and Screening of Wax-deficient Gene Ggl-1 in Garlic (Allium sativum L.) [J]. Biotechnology Bulletin, 2025, 41(9): 219-231. |
| [11] | YAN Meng-yang, LIANG Xiao-yang, DAI Jun-ang, ZHANG Yan, GUAN Tuan, ZHANG Hui, LIU Liang-bo, SUN Zhi-hua. Screening of Amoxicillin-degrading Bacteria and Study on Its Degradation Mechanisms [J]. Biotechnology Bulletin, 2025, 41(9): 314-325. |
| [12] | BAI Yu-guo, LI Wan-di, LIANG Jian-ping, SHI Zhi-yong, LU Geng-long, LIU Hong-jun, NIU Jing-ping. Growth-promoting Mechanism of Trichoderma harzianum T9131 on Astragalus membranaceus Seedlings [J]. Biotechnology Bulletin, 2025, 41(8): 175-185. |
| [13] | ZHANG Yue, BI Yu, MU Xue-nan, ZHENG Zi-wei, WANG Zhi-gang, XU Wei-hui. Biocontrol Characteristics of Strain JB7 against Fusarium graminearum [J]. Biotechnology Bulletin, 2025, 41(7): 261-271. |
| [14] | LI Cheng-hua, DOU Fei-fei, REN Yu-zhao, LIU Cai-xia, LIU Feng-lou, WANG Zhang-jun, LI Qing-feng. Effect of Exogenous Salicylic Acid on Wheat Infested with Blumeria graminis f. sp. tritici and Its Transcriptome Analysis [J]. Biotechnology Bulletin, 2025, 41(7): 272-280. |
| [15] | GUO Xiu-juan, FENG Yu, WU Rui-xiang, WANG Li-qin, YANG Jian-chun. Transcriptome Analysis of the Effect of Ca 2+ Treatment on the Seed Germination of Flax [J]. Biotechnology Bulletin, 2025, 41(7): 139-149. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||