







生物技术通报 ›› 2024, Vol. 40 ›› Issue (9): 212-224.doi: 10.13560/j.cnki.biotech.bull.1985.2024-0276
岳丽昕( ), 王清华, 刘泽洲, 孔素萍, 高莉敏()
), 王清华, 刘泽洲, 孔素萍, 高莉敏()
收稿日期:2024-03-21
出版日期:2024-09-26
发布日期:2024-10-12
通讯作者:
高莉敏,女,硕士,副研究员,研究方向:蔬菜遗传育种;E-mail: lmgao1201@163.com作者简介:岳丽昕,女,博士,助理研究员,研究方向:蔬菜遗传育种;E-mail: yuelixin.happy@163.com
基金资助:
YUE Li-xin(), WANG Qing-hua, LIU Ze-zhou, KONG Su-ping, GAO Li-min()
Received:2024-03-21
Published:2024-09-26
Online:2024-10-12
摘要:
【目的】挖掘大葱雄性不育相关的特异性模块并筛选核心基因,为后续深入解析大葱雄性不育分子机制提供理论依据。【方法】以大葱可育株MF和不育株MS不同发育时期的花蕾为研究对象,基于转录组测序和加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA),鉴定与大葱雄性不育相关的特异性模块,筛选核心基因。【结果】通过WGCNA分析,将8 320个基因划分为18个共表达模块,获得3个与大葱不育株MS花粉败育高度相关的特异性模块(blue、midnightblue和black模块)。功能富集分析表明,大葱不育株MS的花粉败育与苯丙素生物合成、角质和蜡质生物合成、淀粉和蔗糖代谢、α-亚油酸代谢和脂肪酸延长等多种代谢过程密切相关。根据模块内基因的连接度以及基因意义,鉴定得到atpB、TPS9、TPD1、BHLH35等核心基因,可能在大葱花粉发育中起关键调控作用。实时荧光定量PCR(quantitative real-time polymerase chain reaction, RT-qPCR)结果显示,核心基因的表达量在MF和MS不同发育时期的花蕾中差异显著,且表达趋势与转录组学测序结果一致。【结论】鉴定了3个与大葱花粉败育高度相关的特异性模块,筛选到atpB、TPS9、TPD1等大葱花粉发育相关的核心基因,并发现大葱花粉败育主要涉及苯丙素生物合成、角质和蜡质生物合成、淀粉和蔗糖代谢、α-亚油酸代谢和脂肪酸延长等过程。
岳丽昕, 王清华, 刘泽洲, 孔素萍, 高莉敏. 基于转录组和WGCNA筛选大葱雄性不育相关基因[J]. 生物技术通报, 2024, 40(9): 212-224.
YUE Li-xin, WANG Qing-hua, LIU Ze-zhou, KONG Su-ping, GAO Li-min. Screening Genes Related to Male Sterile in Welsh Onion(Allium fistulosum L.) Based on Transcriptomic Profiling and WGCNA[J]. Biotechnology Bulletin, 2024, 40(9): 212-224.
| 基因ID Gene ID | 基因名称Gene name | 正向引物Forward sequence(5'-3') | 反向引物Reverse sequence(5'-3') |
|---|---|---|---|
| AfisC2G08595 | TPS9 | CGCTCTGGTTTGGCATCATCC | ACAACGGCAGGTTCCTTAACAAG |
| AfisC2G08419 | TPD1 | GTTGAAGGCAAACCAGAGTACAGAG | GCTCGACACTGCTTAGACCATAAC |
| AfisC3G06496 | OPR1 | GCCCAAACGCTTTCTCTTAAATGTC | AGCCTGAACCTGAACCTCCTTC |
| AfisC4G05057 | ATPG | ACGAGAGGAATGAGAAGATGAAGTC | AACACGGTAGCAGCAACAGATC |
| AfisC4G00897 | BHLH35 | ATGCCAACTACTGGGAAACCAAGC | TTGTTGTTCCTTCAGGCGAGCTG |
| AfisC7G05890 | APX8 | AGGTGGTGGTGGTGGTTACAG | TCTAGTTCCTCCAGTTTCCATCGG |
| AfisC7G05387 | atpB | CATCAGCGAAATGTTGGATCAAGAC | GTGTTGTTATGCGAACCATTCAGAC |
| Actin | ACACGGCCTGGATAGCAACAT | AGAGCAGTATTCCCAAGCATT |
表1 RT-qPCR引物序列
Table 1 RT-qPCR primers’ sequences
| 基因ID Gene ID | 基因名称Gene name | 正向引物Forward sequence(5'-3') | 反向引物Reverse sequence(5'-3') |
|---|---|---|---|
| AfisC2G08595 | TPS9 | CGCTCTGGTTTGGCATCATCC | ACAACGGCAGGTTCCTTAACAAG |
| AfisC2G08419 | TPD1 | GTTGAAGGCAAACCAGAGTACAGAG | GCTCGACACTGCTTAGACCATAAC |
| AfisC3G06496 | OPR1 | GCCCAAACGCTTTCTCTTAAATGTC | AGCCTGAACCTGAACCTCCTTC |
| AfisC4G05057 | ATPG | ACGAGAGGAATGAGAAGATGAAGTC | AACACGGTAGCAGCAACAGATC |
| AfisC4G00897 | BHLH35 | ATGCCAACTACTGGGAAACCAAGC | TTGTTGTTCCTTCAGGCGAGCTG |
| AfisC7G05890 | APX8 | AGGTGGTGGTGGTGGTTACAG | TCTAGTTCCTCCAGTTTCCATCGG |
| AfisC7G05387 | atpB | CATCAGCGAAATGTTGGATCAAGAC | GTGTTGTTATGCGAACCATTCAGAC |
| Actin | ACACGGCCTGGATAGCAACAT | AGAGCAGTATTCCCAAGCATT |
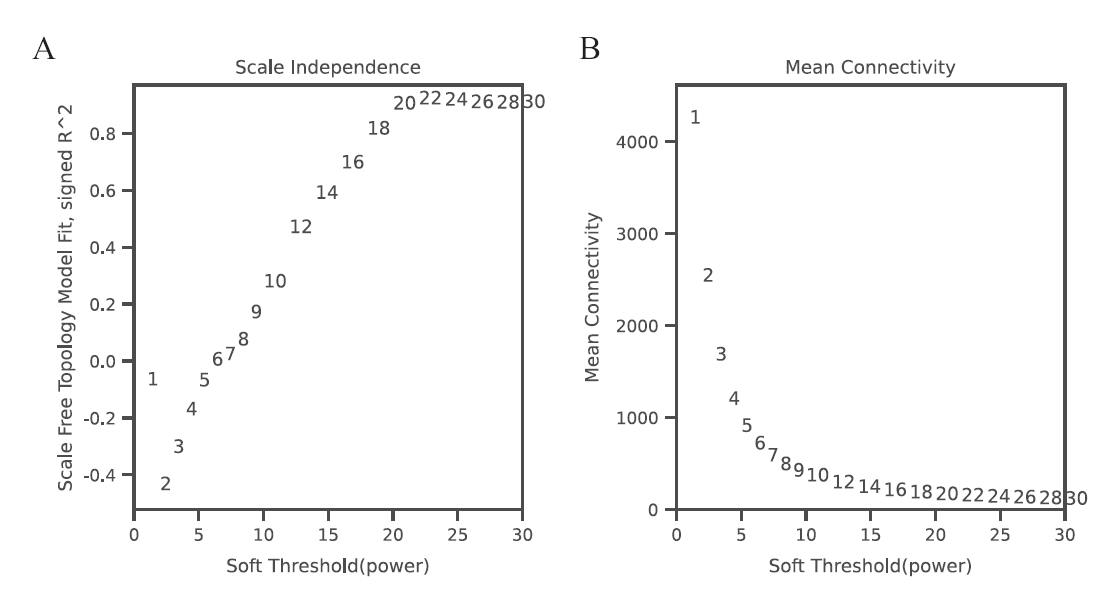
图1 软阈值Power值的确定 A: 无尺度网络模型图;B: 网络平均连接度
Fig. 1 Determination of soft threshold(Power) A: Scale-free network model. B: the average link degree of each soft threshold

图2 层次聚类及模块划分 A: 基因聚类树和模块切割;B: 模块数量
Fig. 2 Hierarchical clustering tree and module divisimg A: Gene cluster dendrograms and module cuting. B: Gene number of each module

图3 模块与花粉发育表型的相关性分析 横坐标上的数字代表不同的花粉发育时期:2代表花粉母细胞时期,3代表四分体时期,4代表单核小孢子时期,5代表二核、三核小孢子时期。字母F和S分别代表可育株MF和不育株MS。红色表示显著正相关,蓝色表示显著负相关
Fig. 3 Correlation analysis between module and anther development phenotype Numbers on the abscissa indicate the various anther development stages: “2” indicates the pollen mother cell stage, “3”indicates the tetrad stage, “4” indicates the uninucleate microspore stage, and “5” indicates the bicellular or tricellular pollen stage. The letters F and S indicate male-fertile(MF)plants and male-sterile(MS)plants, respectively. Red indicates a strong positive correlation and blue indicates a strong negative correlation
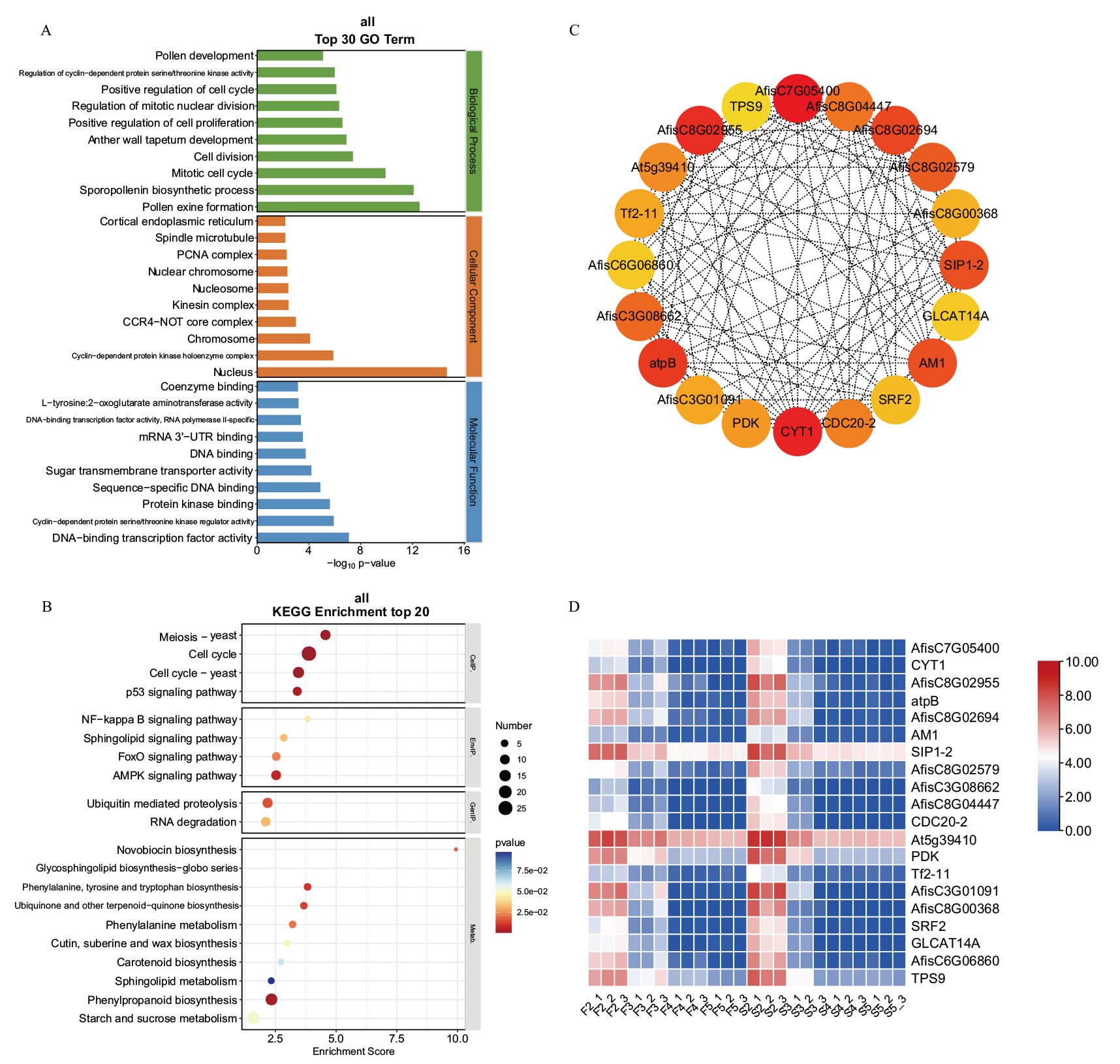
图4 blue模块的功能富集分析及核心基因表达 A: GO富集分析;B: KEGG富集分析;C: 核心基因及其共表达网络;D: 核心基因的表达量热图, 下同
Fig. 4 Functional enrichment analysis and hub gene expression of the blue module A: GO enrichment analysis. B: KEGG enrichment analysis. C: Co-expression gene networks with the greatest hubness in every module. D: Expression profiles of hub genes, the same below
| 基因名称Gene name | 模块 Module | 基因描述 Gene description | GO注释 GO term | KEGG通路 KEGG pathway |
|---|---|---|---|---|
| CYT1 | Blue | 甘露糖-1-磷酸鸟苷酸转移酶1 Mannose-1-phosphate guanylyl transferase 1 | 甘露糖-1-磷酸鸟苷酸转移酶活性Mannose-1-phosphate guanylyl transferase activity GDP-甘露糖生物合成过程GDP-mannose biosynthetic process L-抗坏血酸的生物合成过程L-ascorbic acid biosynthetic process 纤维素生物合成过程Cellulose biosynthetic process | 氨基糖和核苷酸糖代谢 Amino sugar and nucleotide sugar metabolism 果糖和甘露糖代谢途径 Fructose and mannose metabolism |
| TPS9 | Blue | α海藻糖合成酶 9 Alpha-trehalose-phosphate synthase[UDP-forming]9 | 海藻糖生物合成过程Trehalose biosynthetic process 去磷酸化Dephosphorylation 转移酶活性,转移糖基Transferase activity, transferring glycosyl groups 海藻糖代谢对逆境胁迫的响应Trehalose metabolism in response to stress | 淀粉和蔗糖代谢 Starch and sucrose metabolism |
| atpB | Blue | ATP合成酶β亚基 ATP synthase subunit beta | ATP结合ATP binding ATP合成偶联质子转运ATP synthesis coupled proton transport 质子转移ATP合酶复合体,催化核心F(1)Proton-transporting ATP synthase complex, Catalytic core F(1) 质子转移ATP合酶活性,旋转机制Proton-transporting ATP synthase activity, rotational mechanism | 氧化磷酸化 Oxidative phosphorylation |
| TPD1 | Midnightblue | 绒毡层决定因子蛋白 Protein TAPETUM DETERMINANT 1 | 细胞命运决定Cell fate determination 花药发育Anther development | _ |
| ATPG | Midnightblue | ATP合酶亚基b' ATP synthase subunit b' | ATP结合ATP binding 质子跨膜转运蛋白活性Proton transmembrane transporter activity ATP合成偶联质子转运ATP synthesis coupled proton transport 膜的整体成分Integral component of membrane 质子转移ATP合酶复合体,催化核心F(o)Proton-transporting ATP synthase complex, coupling factor F(o) | 氧化磷酸化 Oxidative phosphorylation |
| APX8 | Midnightblue | L-抗坏血酸过氧化物酶8 L-ascorbate peroxidase 8 | 对活性氧的响应Response to reactive oxygen species 过氧化物酶活性Peroxidase activity L-抗坏血酸过氧化物酶活性L-ascorbate peroxidase activity 细胞对氧化应激的反应Cellular response to oxidative stress 过氧化氢分解过程Hydrogen peroxide catabolic process | 谷胱甘肽代谢 Glutathione metabolism |
| OPR1 | Black | 12-氧代二烯酸还原酶1 12-oxophytodienoate reductase 1 | 茉莉酸生物合成过程Jasmonic acid biosynthetic process 12-氧植二烯酸还原酶活性12-oxophytodienoate reductase activity 脂氧化物生物合成过程Oxylipin biosynthetic process | α-亚油酸代谢 Alpha-Linolenic acid metabolism |
| LOX6 | Black | 脂氧合酶6 Lipoxygenase 6 | 茉莉酸生物合成过程Jasmonic acid biosynthetic process 脂氧化物生物合成过程Oxylipin biosynthetic process 脂质氧化Lipid oxidation 亚油酸13S-脂氧合酶活性Linoleate 13S-lipoxygenase activity | 亚油酸代谢 Linoleic acid metabolism |
表2 核心基因的功能注释信息
Table 2 Functional term information of hub genes
| 基因名称Gene name | 模块 Module | 基因描述 Gene description | GO注释 GO term | KEGG通路 KEGG pathway |
|---|---|---|---|---|
| CYT1 | Blue | 甘露糖-1-磷酸鸟苷酸转移酶1 Mannose-1-phosphate guanylyl transferase 1 | 甘露糖-1-磷酸鸟苷酸转移酶活性Mannose-1-phosphate guanylyl transferase activity GDP-甘露糖生物合成过程GDP-mannose biosynthetic process L-抗坏血酸的生物合成过程L-ascorbic acid biosynthetic process 纤维素生物合成过程Cellulose biosynthetic process | 氨基糖和核苷酸糖代谢 Amino sugar and nucleotide sugar metabolism 果糖和甘露糖代谢途径 Fructose and mannose metabolism |
| TPS9 | Blue | α海藻糖合成酶 9 Alpha-trehalose-phosphate synthase[UDP-forming]9 | 海藻糖生物合成过程Trehalose biosynthetic process 去磷酸化Dephosphorylation 转移酶活性,转移糖基Transferase activity, transferring glycosyl groups 海藻糖代谢对逆境胁迫的响应Trehalose metabolism in response to stress | 淀粉和蔗糖代谢 Starch and sucrose metabolism |
| atpB | Blue | ATP合成酶β亚基 ATP synthase subunit beta | ATP结合ATP binding ATP合成偶联质子转运ATP synthesis coupled proton transport 质子转移ATP合酶复合体,催化核心F(1)Proton-transporting ATP synthase complex, Catalytic core F(1) 质子转移ATP合酶活性,旋转机制Proton-transporting ATP synthase activity, rotational mechanism | 氧化磷酸化 Oxidative phosphorylation |
| TPD1 | Midnightblue | 绒毡层决定因子蛋白 Protein TAPETUM DETERMINANT 1 | 细胞命运决定Cell fate determination 花药发育Anther development | _ |
| ATPG | Midnightblue | ATP合酶亚基b' ATP synthase subunit b' | ATP结合ATP binding 质子跨膜转运蛋白活性Proton transmembrane transporter activity ATP合成偶联质子转运ATP synthesis coupled proton transport 膜的整体成分Integral component of membrane 质子转移ATP合酶复合体,催化核心F(o)Proton-transporting ATP synthase complex, coupling factor F(o) | 氧化磷酸化 Oxidative phosphorylation |
| APX8 | Midnightblue | L-抗坏血酸过氧化物酶8 L-ascorbate peroxidase 8 | 对活性氧的响应Response to reactive oxygen species 过氧化物酶活性Peroxidase activity L-抗坏血酸过氧化物酶活性L-ascorbate peroxidase activity 细胞对氧化应激的反应Cellular response to oxidative stress 过氧化氢分解过程Hydrogen peroxide catabolic process | 谷胱甘肽代谢 Glutathione metabolism |
| OPR1 | Black | 12-氧代二烯酸还原酶1 12-oxophytodienoate reductase 1 | 茉莉酸生物合成过程Jasmonic acid biosynthetic process 12-氧植二烯酸还原酶活性12-oxophytodienoate reductase activity 脂氧化物生物合成过程Oxylipin biosynthetic process | α-亚油酸代谢 Alpha-Linolenic acid metabolism |
| LOX6 | Black | 脂氧合酶6 Lipoxygenase 6 | 茉莉酸生物合成过程Jasmonic acid biosynthetic process 脂氧化物生物合成过程Oxylipin biosynthetic process 脂质氧化Lipid oxidation 亚油酸13S-脂氧合酶活性Linoleate 13S-lipoxygenase activity | 亚油酸代谢 Linoleic acid metabolism |
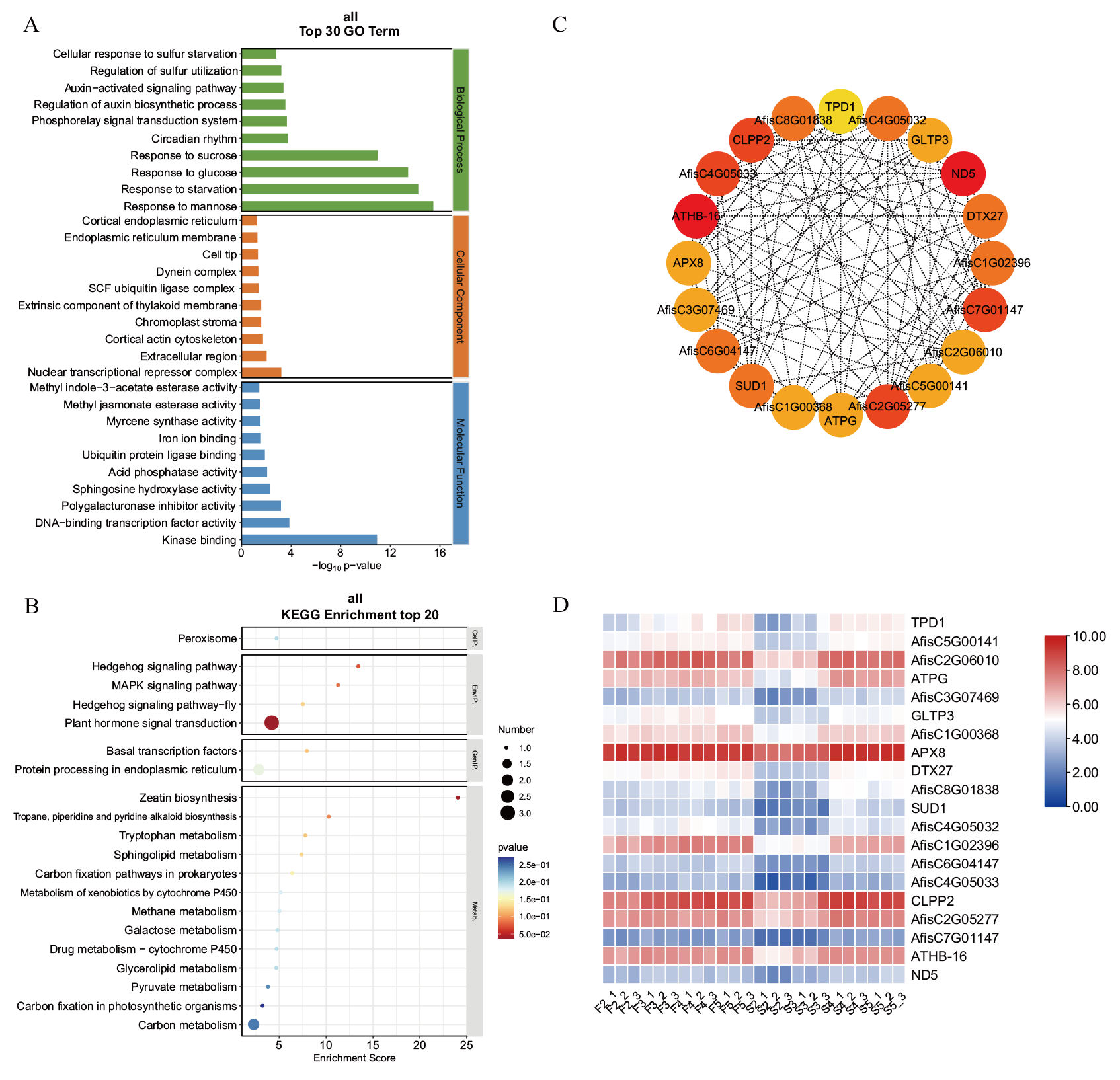
图5 Midnightblue模块的功能富集分析及核心基因表达
Fig. 5 Functional enrichment analysis and hub gene expression of the midnightblue module

图6 Black模块的功能富集分析及核心基因表达
Fig. 6 Functional enrichment analysis and hub gene expression of the Black module
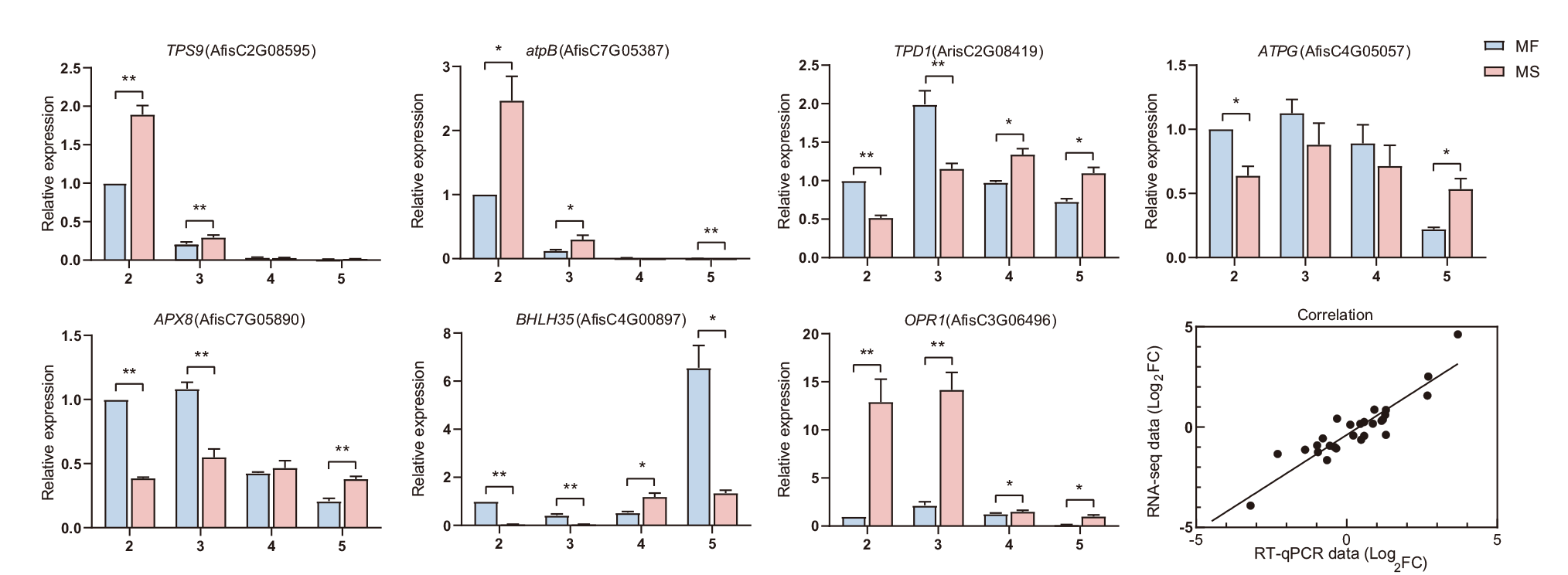
图7 7个核心基因的RT-qPCR验证 横坐标上的数字代表不同的花粉发育时期:2代表花粉母细胞时期,3代表四分体时期,4代表单核小孢子时期,5代表二核、三核小孢子时期。蓝色条为可育株MF,粉色条为不育株MS。*:统计显著性差异(t检验,P < 0.05);**:极显著性差异(P < 0.01)
Fig. 7 RT-qPCR analysis of seven hub genes Numbers on the abscissa indicate the various anther development stages: “2” indicates the pollen mother cell stage, “3” indicates the tetrad stage, “4” indicates the uninucleate microspore stage, and “5” indicates the bicellular or tricellular pollen stage. Blue bars indicate MF plants and pink bars indicate MS plants. ‘*’ indicates statistically significant differences(t-test, P < 0.05), and ‘**’ indicates extremely significant differences(P < 0.01)
| [1] | 乔立娟, 吴曼, 宗义湘, 等. 我国辛辣类蔬菜产业发展趋势与建议[J]. 中国蔬菜, 2021(8): 11-17. |
| Qiao LJ, Wu M, Zong YX, et al. Development trend and suggestions of spicy vegetable industry in China[J]. China Veg, 2021(8): 11-17. | |
| [2] | 霍晴, 吴曼, 赵邦宏. 我国大葱产业竞争力分析与对策[J]. 中国蔬菜, 2022(3): 1-8. |
| Huo Q, Wu M, Zhao BH. Analysis and countermeasures on the competitiveness of Chinese Welsh onion industry[J]. China Veg, 2022(3): 1-8. | |
| [3] | 岳丽昕, 王清华, 刘泽洲, 等. 大葱雄性不育花药败育的形态学特征和细胞学研究[J]. 中国蔬菜, 2023(9): 58-68. |
| Yue LX, Wang QH, Liu ZZ, et al. Morphological characteristics of Welsh onion(Allium fistulosum L.) male sterile anther abortion and studies on cytology[J]. China Veg, 2023(9): 58-68. | |
| [4] | Padula G, Xia XZ, Hołubowicz R. Welsh onion(Allium fistulosum L.) seed physiology, breeding, production and trade[J]. Plants, 2022, 11(3): 343. |
| [5] | 康香辉, 安进军, 袁瑞江, 等. 大葱雄性不育的研究和应用进展[J]. 长江蔬菜, 2021(4): 43-47. |
| Kang XH, An JJ, Yuan RJ, et al. Research progress and application of male sterility in Welsh onion(Allium fistulosum L. var. gigantum makino)[J]. J Changjiang Veg, 2021(4): 43-47. | |
| [6] | Liu QC, Lan YP, Wen CL, et al. Transcriptome sequencing analyses between the cytoplasmic male sterile line and its maintainer line in Welsh onion(Allium fistulosum L.)[J]. Int J Mol Sci, 2016, 17(7): 1058. |
| [7] | Gao LM, Huo YM, Chen W, et al. Identification of allelic variation in the atp6 gene and its use in developing molecular markers for distinguishing two cytoplasmic types of bunching onion(Allium fistulosum L.)[J]. J Hortic Sci Biotechnol, 2018, 93(5): 450-455. |
| [8] | Gao LM, Chen YQ, Huo YM, et al. Development of SCAR markers to distinguish male-sterile and normal cytoplasm in bunching onion(Allium fistulosum L.)[J]. J Hortic Sci Biotechnol, 2015, 90(1): 57-62. |
| [9] | Wang C, Li HY, Zhang LY, et al. Identification of an AFLP marker and conversion to a SCAR marker to identify cytoplasmic male-sterile or normal cytoplasm in Welsh onion(Allium fistulosum L.)[J]. J Hortic Sci Biotechnol, 2013, 88(4): 409-414. |
| [10] | 盖树鹏, 孟祥栋. 大葱(Allium fistulosum L.)胞质雄性不育基因的RAPD标记[J]. 农业生物技术学报, 2002, 10(1): 94-97. |
| Gai SP, Meng XD. Development and identification RAPD markers linked to cytoplasmic male sterilityin Welsh onion(Allium fistulosum L.)[J]. J Agric Biotechnol, 2002, 10(1): 94-97. | |
| [11] | 高莉敏, 董飞, 霍雨猛, 等. 大葱细胞质雄性不育基因的SCAR标记开发[J]. 园艺学报, 2013, 40(7): 1382-1388. |
| Gao LM, Dong F, Huo YM, et al. Development of SCAR marker identifying the cytoplasmic male sterility gene in bunching onion(Allium fistulosum L.)[J]. Acta Hortic Sin, 2013, 40(7): 1382-1388. | |
| [12] | Liu QC, Wen CL, Zhao H, et al. Comparative analysis of male sterility associated ATPase isoenzymes and atpA genes in a Welsh onion(Allium fistulosum L.) cytoplasmic male sterility line and its maintainer line[J]. Sci Hortic, 2019, 243: 101-106. |
| [13] | 王清华, 张荣亭, 贾述娟, 等. 大葱雄性不育系及保持系atp6基因的克隆及表达分析[J]. 分子植物育种, 2021, 19(7): 2163-2168. |
| Wang QH, Zhang RT, Jia SJ, et al. Cloning and expression analysis of atp6 genes between cytoplasmic male sterile line and maintainer line in Welsh onion[J]. Mol Plant Breed, 2021, 19(7): 2163-2168. | |
| [14] | Liao NQ, Hu ZY, Miao JS, et al. Chromosome-level genome assembly of bunching onion illuminates genome evolution and flavor formation in Allium crops[J]. Nat Commun, 2022, 13(1): 6690. |
| [15] | Hao F, Liu X, Zhou BT, et al. Chromosome-level genomes of three key Allium crops and their trait evolution[J]. Nat Genet, 2023, 55(11): 1976-1986. |
| [16] |
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis[J]. BMC Bioinformatics, 2008, 9: 559.
doi: 10.1186/1471-2105-9-559 pmid: 19114008 |
| [17] |
Roberts WR, Roalson EH. Comparative transcriptome analyses of flower development in four species of Achimenes(Gesneriaceae)[J]. BMC Genomics, 2017, 18(1): 240.
doi: 10.1186/s12864-017-3623-8 pmid: 28320315 |
| [18] | Li YQ, Qin TF, Wei CY, et al. Using transcriptome analysis to screen for key genes and pathways related to cytoplasmic male sterility in cotton(Gossypium hirsutum L.)[J]. Int J Mol Sci, 2019, 20(20): 5120. |
| [19] | Zhang TB, Yuan SH, Liu ZH, et al. Comparative transcriptome analysis reveals hormone signal transduction and sucrose metabolism related genes involved in the regulation of anther dehiscence in photo-thermo-sensitive genic male sterile wheat[J]. Biomolecules, 2022, 12(8): 1149. |
| [20] | 王志敏, 袁超, 丁泽琴, 等. 功能雄性不育茄子差异基因及代谢途径分析[J]. 生物工程学报, 2021, 37(1): 253-265. |
| Wang ZM, Yuan C, Ding ZQ, et al. Analysis of differential genes and metabolic pathway related to functional male sterility in eggplant[J]. Chin J Biotechnol, 2021, 37(1): 253-265. | |
| [21] | Liu YZ, Sun H, Ye R, et al. Potential candidate genes and pathways related to cytoplasmic male sterility in Dianthus spiculifolius as revealed by transcriptome analysis[J]. Plant Cell Rep, 2023, 42(9): 1503-1516. |
| [22] | Chen SF, Zhou YQ, Chen YR, et al. Fastp: an ultra-fast all-in-one FASTQ preprocessor[J]. Bioinformatics, 2018, 34(17): i884-i890. |
| [23] |
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements[J]. Nat Methods, 2015, 12(4): 357-360.
doi: 10.1038/nmeth.3317 pmid: 25751142 |
| [24] | Roberts A, Trapnell C, Donaghey J, et al. Improving RNA-Seq expression estimates by correcting for fragment bias[J]. Genome Biol, 2011, 12(3): R22. |
| [25] | Yue LX, Li GL, Dai Y, et al. Gene co-expression network analysis of the heat-responsive core transcriptome identifies hub genes in Brassica rapa[J]. Planta, 2021, 253(5): 111. |
| [26] | Chin CH, Chen SH, Wu HH, et al. cytoHubba: identifying hub objects and sub-networks from complex interactome[J]. BMC Syst Biol, 2014, 8(Suppl 4): S11. |
| [27] | Chen CJ, Wu Y, Li JW, et al. TBtools-II: a “one for all, all for one” bioinformatics platform for biological big-data mining[J]. Mol Plant, 2023, 16(11): 1733-1742. |
| [28] |
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T))method[J]. Methods, 2001, 25(4): 402-408.
doi: 10.1006/meth.2001.1262 pmid: 11846609 |
| [29] | Han YC, Gao YJ, Li Y, et al. Chloroplast genes are involved in the male-sterility of K-type CMS in wheat[J]. Genes, 2022, 13(2): 310. |
| [30] | Zhong XH, Chen DH, Cui J, et al. Comparative analysis of the complete mitochondrial genome sequences and anther development cytology between maintainer and Ogura-type cytoplasm male-sterile cabbage(B. oleracea Var. capitata)[J]. BMC Genomics, 2021, 22(1): 646. |
| [31] | Palumbo F, Vitulo N, Vannozzi A, et al. The mitochondrial genome assembly of fennel(Foeniculum vulgare)reveals two different atp6 gene sequences in cytoplasmic male sterile accessions[J]. Int J Mol Sci, 2020, 21(13): 4664. |
| [32] |
Kong XJ, Liu DM, Zheng J, et al. RNA editing analysis of ATP synthase genes in the cotton cytoplasmic male sterile line H276A[J]. Biol Res, 2019, 52(1): 6.
doi: 10.1186/s40659-019-0212-0 pmid: 30728078 |
| [33] |
Tang DF, Wei F, Zhou RY. Comparative analysis of chloroplast genomes of kenaf cytoplasmic male sterile line and its maintainer line[J]. Sci Rep, 2021, 11(1): 5301.
doi: 10.1038/s41598-021-84567-1 pmid: 33674697 |
| [34] | Du K, Xiao YY, Liu QE, et al. Abnormal tapetum development and energy metabolism associated with sterility in SaNa-1A CMS of Brassica napus L[J]. Plant Cell Rep, 2019, 38(5): 545-558. |
| [35] | Li N, Zhang DS, Liu HS, et al. The rice tapetum degeneration retardation gene is required for tapetum degradation and anther development[J]. Plant Cell, 2006, 18(11): 2999-3014. |
| [36] | 陈玮玥. BES1家族转录因子调控拟南芥花药绒毡层的发育[D]. 兰州: 兰州大学, 2022. |
| Chen WY. BES1 family transcription factors regulate Arabidopsis tapetum development[D]. Lanzhou: Lanzhou University, 2022. | |
| [37] | Huang J, Zhang TY, Linstroth L, et al. Control of anther cell differentiation by the small protein ligand TPD1 and its receptor EMS1 in Arabidopsis[J]. PLoS Genet, 2016, 12(8): e1006147. |
| [38] | 陈广霞. 拟南芥TPD1-EMS1信号转导途径相关基因的鉴定与分析[D]. 北京: 中国农业大学, 2015. |
| Chen GX. Identification and analysis of the genes related to TPD1-EMS1 signaling pathway in Arabidopsis[D]. Beijing: China Agricultural University, 2015. | |
| [39] | Yang SL, Jiang LX, Puah CS, et al. Overexpression of TAPETUM DETERMINANT1 alters the cell fates in the Arabidopsis carpel and tapetum via genetic interaction with excess microsporocytes1/extra sporogenous cells[J]. Plant Physiol, 2005, 139(1): 186-191. |
| [40] | Hu CH, Sheng O, Dong T, et al. Overexpression of MaTPD1A impairs fruit and pollen development by modulating some regulators in Musa itinerans[J]. BMC Plant Biol, 2020, 20(1): 402. |
| [41] | Han FQ, Yuan KW, Kong CC, et al. Fine mapping and candidate gene identification of the genic male-sterile gene ms3 in cabbage 51S[J]. Theor Appl Genet, 2018, 131(12): 2651-2661. |
| [42] | Sun YJ, Fu M, Ang YN, et al. Combined analysis of transcriptome and metabolome reveals that sugar, lipid, and phenylpropane metabolism are essential for male fertility in temperature-induced male sterile rice[J]. Front Plant Sci, 2022, 13: 945105. |
| [43] |
Lastdrager J, Hanson J, Smeekens S. Sugar signals and the control of plant growth and development[J]. J Exp Bot, 2014, 65(3): 799-807.
doi: 10.1093/jxb/ert474 pmid: 24453229 |
| [44] | Hao MM, Yang WL, Li TD, et al. Combined transcriptome and proteome analysis of anthers of AL-type cytoplasmic male sterile line and its maintainer line reveals new insights into mechanism of male sterility in common wheat[J]. Front Genet, 2021, 12: 762332. |
| [45] | 常小瑶. 基于转录组和代谢组解析地梢瓜花药发育的调控机制[D]. 呼和浩特: 内蒙古农业大学, 2023. |
| Chang XY. Molecular mechanism of anther development in Cynanchum thesioides based on transcriptome and metabolomic[D]. Hohhot: Inner Mongolia Agricultural University, 2023. | |
| [46] | 刘永明, 张玲, 周建瑜, 等. 植物细胞核雄性不育相关bHLH转录因子研究进展[J]. 遗传, 2015, 37(12): 1194-1203. |
| Liu YM, Zhang L, Zhou JY, et al. Research progress of the bHLH transcription factors involved in genic male sterility in plants[J]. Hereditas, 2015, 37(12): 1194-1203. | |
| [47] | Sorensen AM, Kröber S, Unte US, et al. The Arabidopsis ABORTED MICROSPORES(AMS)gene encodes a MYC class transcription factor[J]. Plant J, 2003, 33(2): 413-423. |
| [48] | 李紫良. 小麦温光敏不育系BNS中乙烯响应因子基因TaERF7的功能分析[D]. 杨凌: 西北农林科技大学, 2020. |
| Li ZL. Functional analysis of ethylene response factor gene TaERF7 in thermo-photo-sensitive male sterile of BNS in wheat[D]. Yangling: Northwest A & F University, 2020. | |
| [49] | 李春. 十字花科植物白菜花粉败育过程及其影响机理研究[D]. 昆明: 云南大学, 2022. |
| Li C. Research on the pollen abortion and its underlying mechanisms of Brassica rapa[D]. Kunming: Yunnan University, 2022. |
| [1] | 赵海平, 刘林, 王昕璐, 岳鹏飞, 孔维军, 王蒙. 基于WGCNA探讨葛根素对呕吐毒素诱导C6细胞损伤的保护作用[J]. 生物技术通报, 2024, 40(9): 301-310. |
| [2] | 高萌萌, 赵天宇, 焦馨悦, 林春晶, 关哲允, 丁孝羊, 孙妍妍, 张春宝. 大豆细胞质雄性不育系及其恢复系的比较转录组分析[J]. 生物技术通报, 2024, 40(7): 137-149. |
| [3] | 廖杨梅, 赵国春, 翁学煌, 贾黎明, 陈仲. 无患子雄性不育品种‘琦蕊’不同发育时期雄花转录组分析[J]. 生物技术通报, 2024, 40(7): 197-206. |
| [4] | 白志元, 徐菲, 杨午, 王明贵, 杨玉花, 张海平, 张瑞军. 大豆细胞质雄性不育弱恢复型杂种F1育性转变的转录组分析[J]. 生物技术通报, 2024, 40(6): 134-142. |
| [5] | 秦健, 李振月, 何浪, 李俊玲, 张昊, 杜荣. 肌源性细胞分化的单细胞转录谱变化及细胞间通讯分析[J]. 生物技术通报, 2024, 40(6): 330-342. |
| [6] | 张震, 李清, 徐菁, 陈凯园, 张春芝, 祝光涛. 马铃薯线粒体靶向表达载体的构建与应用[J]. 生物技术通报, 2024, 40(5): 66-73. |
| [7] | 杨淇, 魏子迪, 宋娟, 童堃, 杨柳, 王佳涵, 刘海燕, 栾维江, 马轩. 水稻组蛋白H1三突变体的创建和转录组学分析[J]. 生物技术通报, 2024, 40(4): 85-96. |
| [8] | 赵锐, 狄靖宜, 张广通, 刘浩, 高伟霞. 基于转录组学挖掘兽疫链球菌内源表达元件及高产透明质酸应用[J]. 生物技术通报, 2024, 40(10): 296-304. |
| [9] | 赵金玲, 安磊, 任晓亮. 单细胞转录组测序技术及其在秀丽隐杆线虫中的应用[J]. 生物技术通报, 2023, 39(6): 158-170. |
| [10] | 熊和丽, 沙茜, 刘韶娜, 相德才, 张斌, 赵智勇. 单细胞转录组测序技术在动物上的应用研究[J]. 生物技术通报, 2022, 38(3): 226-233. |
| [11] | 寇佳怡, 王玉玲, 曾睿琳, 兰道亮. 单细胞转录组测序技术及在哺乳动物上的应用[J]. 生物技术通报, 2022, 38(11): 41-48. |
| [12] | 郑青波, 叶娜, 张哓兰, 包鹏甲, 王福彬, 任稳稳, 廖月姣, 阎萍, 潘和平. 天祝白牦牛退行期毛囊细胞亚群鉴定以及特征基因生物信息学分析[J]. 生物技术通报, 2022, 38(10): 262-272. |
| [13] | 洪军, 卫夏怡, 吉冰洁, 叶延欣, 程天赐. 铜绿假单胞菌对鲎素耐药前后的差异表达基因及SNP变化研究[J]. 生物技术通报, 2021, 37(9): 191-202. |
| [14] | 陈建军, 赵怡迪, 曹香林. 脂多糖对鲤肠上皮细胞转录组模式的调控分析[J]. 生物技术通报, 2021, 37(8): 213-220. |
| [15] | 叶娜, 张晓兰, 包鹏甲, 王兴东, 阎萍, 潘和平. 单细胞测序技术及其在毛囊发育中的应用[J]. 生物技术通报, 2021, 37(10): 245-256. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||