







生物技术通报 ›› 2024, Vol. 40 ›› Issue (7): 197-206.doi: 10.13560/j.cnki.biotech.bull.1985.2024-0039
廖杨梅1( ), 赵国春1, 翁学煌2, 贾黎明1, 陈仲1()
), 赵国春1, 翁学煌2, 贾黎明1, 陈仲1()
收稿日期:2024-01-12
出版日期:2024-07-26
发布日期:2024-05-24
通讯作者:
陈仲,男,博士,副教授,研究方向:林木栽培生理及功能基因组学;E-mail: zhongchen@bjfu.edu.cn作者简介:廖杨梅,女,硕士研究生,研究方向:用材林与能源林培育理论与技术;E-mail: 2414729525@qq.com
基金资助:
LIAO Yang-mei1(), ZHAO Guo-chun1, WENG Xue-huang2, JIA Li-ming1, CHEN Zhong1()
Received:2024-01-12
Published:2024-07-26
Online:2024-05-24
摘要:
【目的】 探究无患子(Sapindus mukorossi Gaertn.)雄性不育品种‘琦蕊’雄花不同发育时期的细胞学特征及差异表达基因,为深入解析无患子雄性不育发生的分子机制提供理论依据。【方法】 在观察雄花花药不同发育过程的细胞学特点基础上,进一步利用RNA-Seq技术分别对小孢子母细胞时期(T1)、四分体时期(T2)、单核小孢子时期(T3)的雄花进行转录组比较分析,筛选关键差异表达基因。【结果】 ‘琦蕊’绒毡层和内层细胞的持续膨大与增殖使得药室结构混乱,药室空间不足,最终导致无可育花粉产生。内源激素测定发现,茉莉酸水平在‘琦蕊’雄花发育过程中呈下降趋势。3个时期的转录组分析共筛选到2 990个差异表达基因(differentially expressed genes, DEGs),其中T2_vs_T1中722个(516个上调,206个下调),T3_vs_T2中1 741个(765个上调,976个下调)。GO和KEGG分析表明,这些DEGs主要富集在代谢过程、细胞分裂、花粉壁合成、激素信号转导等方面,共鉴定到32个花粉发育相关DEGs、27个激素合成及信号转导DEGs。随机选取9个DEGs进行RT-qPCR分析,结果与RNA-Seq数据趋势一致,证明转录组数据的准确性。【结论】 绒毡层细胞持续增殖和内层细胞延迟退化不断挤压小孢子是导致无患子‘琦蕊’雄性不育发生的主要原因,物质代谢、细胞分裂、激素水平、花粉发育等相关基因在雄性不育发生过程中发挥重要作用。
廖杨梅, 赵国春, 翁学煌, 贾黎明, 陈仲. 无患子雄性不育品种‘琦蕊’不同发育时期雄花转录组分析[J]. 生物技术通报, 2024, 40(7): 197-206.
LIAO Yang-mei, ZHAO Guo-chun, WENG Xue-huang, JIA Li-ming, CHEN Zhong. Transcriptome Sequencing of Male Sterile Buds at Different Developmental Stages in Sapindus mukorossi ‘Qirui’[J]. Biotechnology Bulletin, 2024, 40(7): 197-206.
| 基因编号Gene ID | 基因名称Gene name | 正向引物序列Forward primer sequence(5'-3') | 反向引物序列Reverse primer sequence(5'-3') |
|---|---|---|---|
| Samuk01G0221000 | SmDYT1 | CACTTACATAGAGGTGCTG | CTCAATCCCACTATTCTTC |
| Samuk09G0006500 | SmMYB35 | ACATACCTCACCACCACCT | TTGACCATCTTCTGCCTAT |
| Samuk03G0239600 | SmbHLH091 | ACGGCAGGAAGATGTCAGT | GTCGTGAATCAAATGGGTG |
| Samuk02G0196100 | SmCAT2 | AACCCAAAGTCTCACATCC | CACCCACATCATCAAAGAG |
| Samuk12G0082500 | SmMIOX1 | TGCTGAGGCTATTAGGAAG | CGAAGTTAGGCAGAAGAAG |
| Samuk07G0073400 | SmCYP704B1 | AAAAGCAGGAGGGATGGTG | TGTATGAGGCTGCGTCAGG |
| Samuk07G0100300 | SmMYB62 | AACAGCCAAATCACAGACG | GCTCCTAGATTGAACCCAC |
| Samuk03G0033700 | SmTCF1 | TTAAGTCCAATATGCGTGTC | GCCCAAACTGATTCCCAC |
| Samuk04G0015800 | SmCER1 | GGCATCCAGGTTACCACAT | CATCTCCCACCACCCATAC |
| Samuk07G0011500 | SmRPL1 | CTGACCACCCCACCACTCTC | TCCTCCTCATCTGCCACCTC |
表1 RT-qPCR引物信息
Table 1 Primers used in the RT-qPCR
| 基因编号Gene ID | 基因名称Gene name | 正向引物序列Forward primer sequence(5'-3') | 反向引物序列Reverse primer sequence(5'-3') |
|---|---|---|---|
| Samuk01G0221000 | SmDYT1 | CACTTACATAGAGGTGCTG | CTCAATCCCACTATTCTTC |
| Samuk09G0006500 | SmMYB35 | ACATACCTCACCACCACCT | TTGACCATCTTCTGCCTAT |
| Samuk03G0239600 | SmbHLH091 | ACGGCAGGAAGATGTCAGT | GTCGTGAATCAAATGGGTG |
| Samuk02G0196100 | SmCAT2 | AACCCAAAGTCTCACATCC | CACCCACATCATCAAAGAG |
| Samuk12G0082500 | SmMIOX1 | TGCTGAGGCTATTAGGAAG | CGAAGTTAGGCAGAAGAAG |
| Samuk07G0073400 | SmCYP704B1 | AAAAGCAGGAGGGATGGTG | TGTATGAGGCTGCGTCAGG |
| Samuk07G0100300 | SmMYB62 | AACAGCCAAATCACAGACG | GCTCCTAGATTGAACCCAC |
| Samuk03G0033700 | SmTCF1 | TTAAGTCCAATATGCGTGTC | GCCCAAACTGATTCCCAC |
| Samuk04G0015800 | SmCER1 | GGCATCCAGGTTACCACAT | CATCTCCCACCACCCATAC |
| Samuk07G0011500 | SmRPL1 | CTGACCACCCCACCACTCTC | TCCTCCTCATCTGCCACCTC |

图1 ‘琦蕊’花药早期发育显微结构特征 T1、T2、T3分别指无患子花发育的第8期(S8)、第9期(S9)、第10b期(S10b),S12为无患子花发育的第12期;T:绒毡层;Tds:四分体;E:表皮;En:内层;ML:中层;比例尺=100 μm。下同
Fig. 1 Microstructure of early development of ‘Qirui’ anther T1, T2 and T3 refer to the 8th(S8), 9th(S9)and 10b(S10b)stages of S. mukorossi Gaertn. flower development respectively, and S12 refers to the 12th stage. T: Tapetum; Tds: tetrad; E: epidermis; En: endothecium; ML: middle layer. Bar = 100 μm. The same below
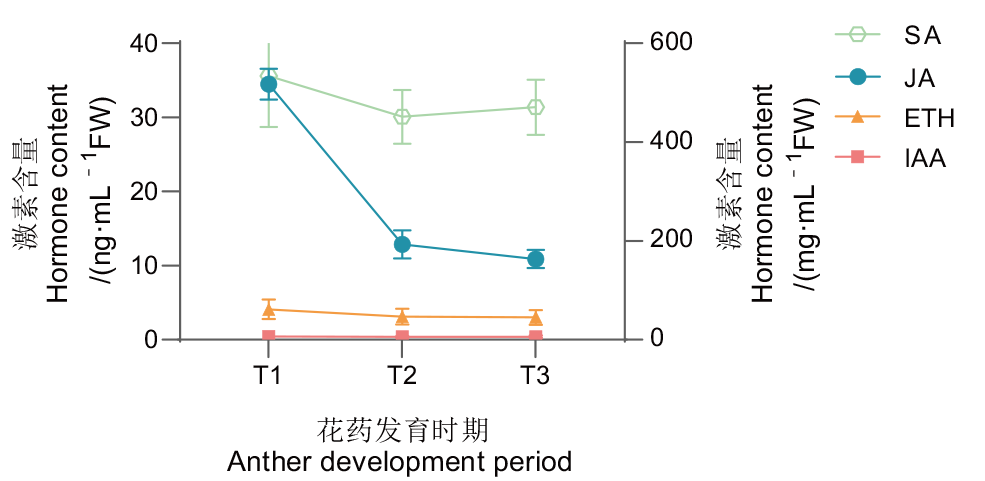
图2 ‘琦蕊’雄花发育过程内源激素含量变化 JA:茉莉酸;SA:水杨酸;IAA:生长素;ET:乙烯
Fig. 2 Changes of endogenous hormone content during male buds development of ‘Qirui’ JA: Jasmonic acid; SA:salicylic acid; IAA: auxin; ETH: ethylene
| 样本名称 Sample name | 原始数据 Raw reads | 过滤后数据 Total clean reads | 总映射率 Total mapping ratio/% | 单一映射率 Uniq mapped reads/% | Q20/% | Q30/% |
|---|---|---|---|---|---|---|
| T1-1 | 88 252 858 | 85 911 910 | 95.86 | 72.67 | 98.37 | 95.02 |
| T1-2 | 91 157 116 | 88 898 424 | 95.83 | 82.08 | 98.46 | 95.22 |
| T1-3 | 88 984 514 | 86 405 806 | 95.85 | 75.65 | 98.10 | 94.31 |
| T2-1 | 88 937 894 | 86 784 106 | 95.91 | 80.89 | 98.28 | 94.76 |
| T2-2 | 100 745 016 | 98 376 588 | 95.87 | 82.46 | 98.42 | 95.11 |
| T2-3 | 107 473 734 | 105 653 102 | 95.97 | 84.99 | 98.44 | 95.12 |
| T3-1 | 95 388 566 | 93 391 938 | 95.77 | 86.11 | 98.36 | 94.95 |
| T3-2 | 89 881 480 | 87 909 282 | 96.08 | 80.18 | 98.43 | 95.17 |
| T3-3 | 96 582 048 | 95 072 204 | 96.00 | 84.08 | 98.39 | 95.00 |
表2 转录组测序数据评估
Table 2 Data evaluation of sample sequencing
| 样本名称 Sample name | 原始数据 Raw reads | 过滤后数据 Total clean reads | 总映射率 Total mapping ratio/% | 单一映射率 Uniq mapped reads/% | Q20/% | Q30/% |
|---|---|---|---|---|---|---|
| T1-1 | 88 252 858 | 85 911 910 | 95.86 | 72.67 | 98.37 | 95.02 |
| T1-2 | 91 157 116 | 88 898 424 | 95.83 | 82.08 | 98.46 | 95.22 |
| T1-3 | 88 984 514 | 86 405 806 | 95.85 | 75.65 | 98.10 | 94.31 |
| T2-1 | 88 937 894 | 86 784 106 | 95.91 | 80.89 | 98.28 | 94.76 |
| T2-2 | 100 745 016 | 98 376 588 | 95.87 | 82.46 | 98.42 | 95.11 |
| T2-3 | 107 473 734 | 105 653 102 | 95.97 | 84.99 | 98.44 | 95.12 |
| T3-1 | 95 388 566 | 93 391 938 | 95.77 | 86.11 | 98.36 | 94.95 |
| T3-2 | 89 881 480 | 87 909 282 | 96.08 | 80.18 | 98.43 | 95.17 |
| T3-3 | 96 582 048 | 95 072 204 | 96.00 | 84.08 | 98.39 | 95.00 |
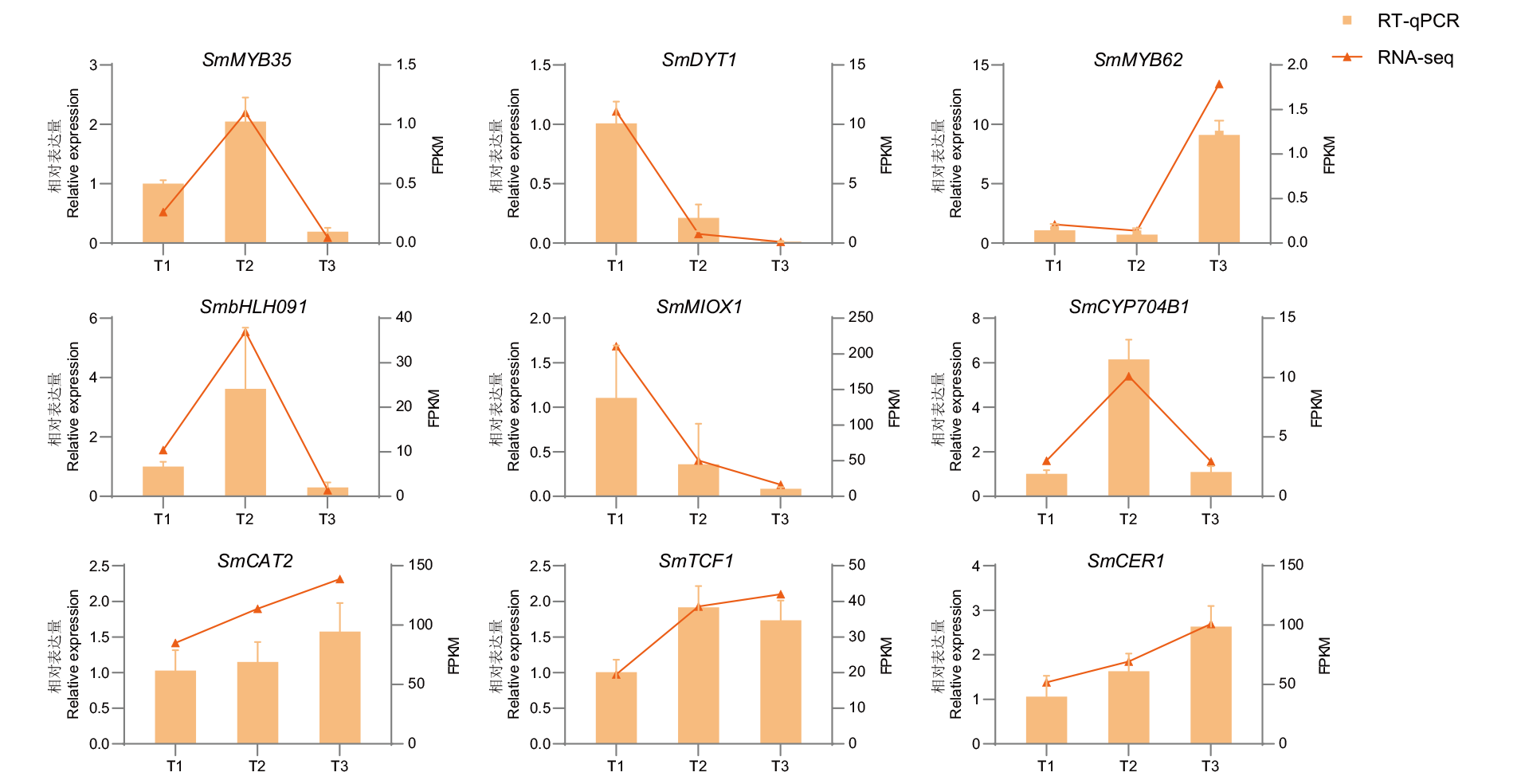
图3 差异表达基因的RT-qPCR验证
Fig. 3 RT-qPCR verification of differentially expressed genes
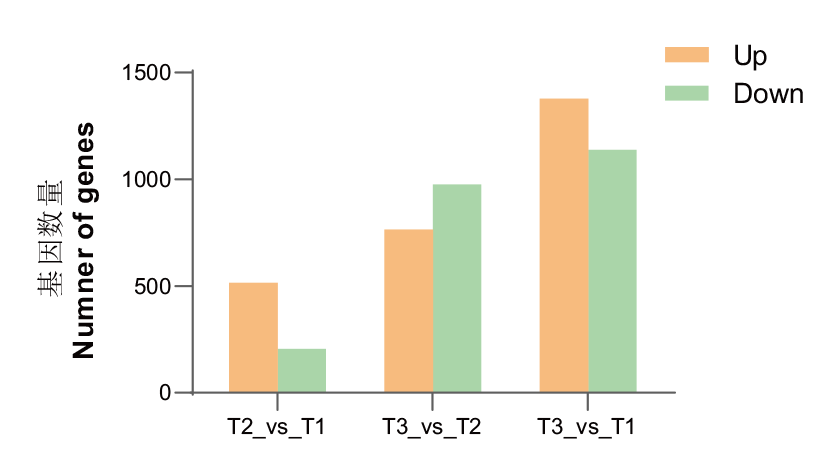
图4 不同时期的差异表达基因
Fig. 4 Numbers of differentially expressed genes in different periods
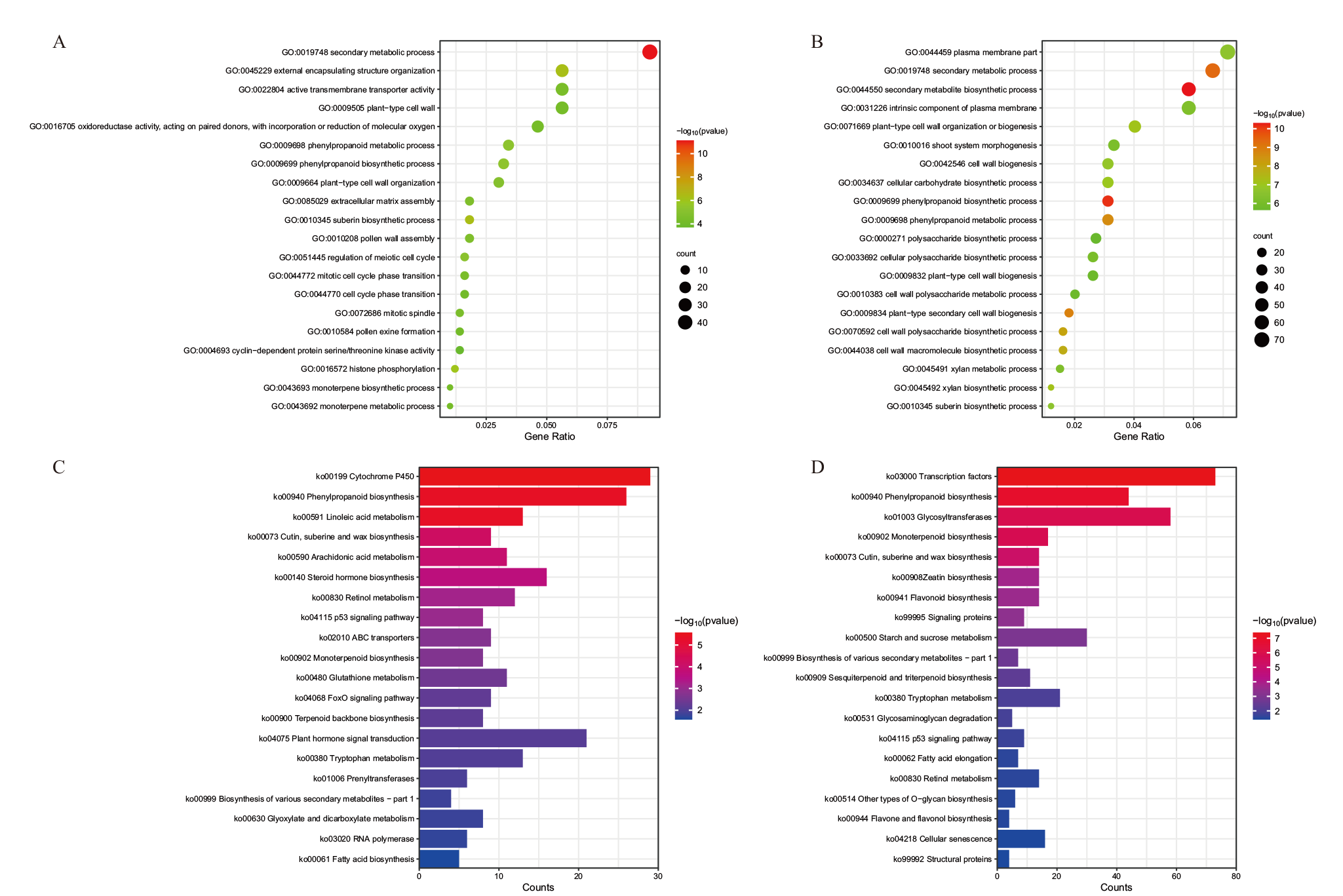
图5 差异表达基因GO功能富集和KEGG通路富集分析 A、C:T2_vs_T1中差异表达基因的GO功能注释(A)和KEGG通路富集(C);B、D:T3_vs_T2中差异表达基因的GO功能注释(B)和KEGG通路富集(D)
Fig. 5 GO functional enrichment and KEGG pathway enrichment of differentially expressed genes A, C: GO functional annotation(A)and KEGG pathway enrichment(C)of differentially expressed genes in T2_vs_T1; B, D: GO functional annotation(B)and KEGG pathway enrichment(D)of differentially expressed genes in T3_vs_T2
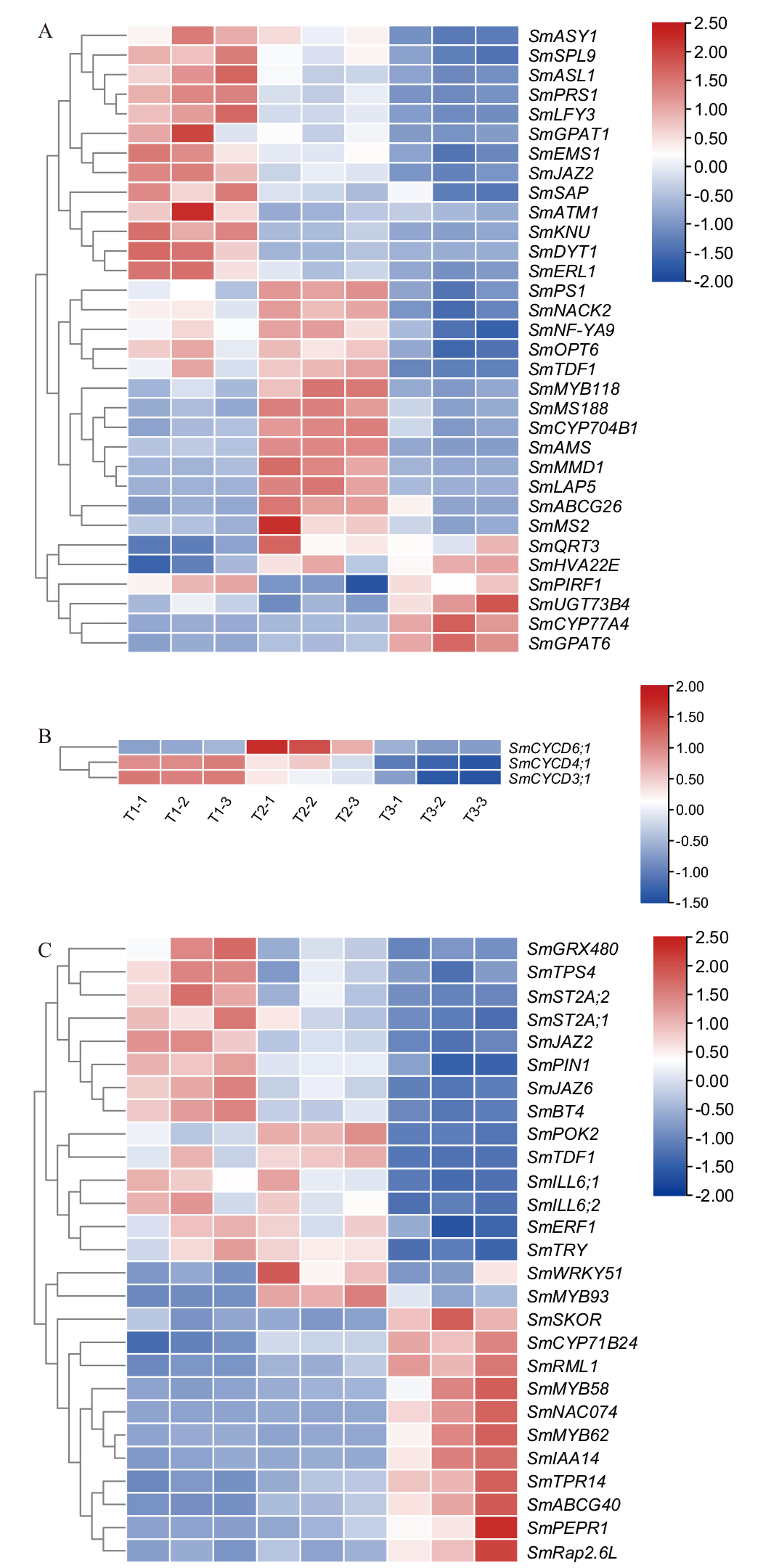
图6 雄性不育关键候选差异基因表达分析 A:花粉发育基因;B:细胞分裂CYCD家族3个基因;C:茉莉酸合成及信号转导基因
Fig. 6 Expression analysis of key candidate differential genes for male sterility A: Pollen development genes. B: Cell division CYCD family of 3 genes. C: Jasmonic acid synthesis and signal transduction genes
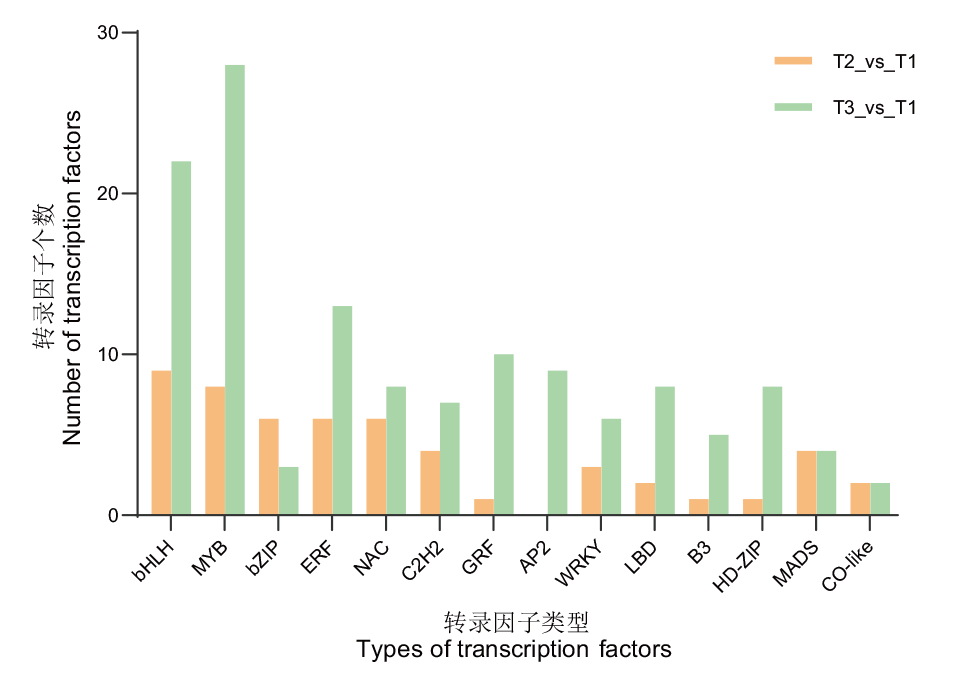
图7 差异表达转录因子家族分析
Fig. 7 Analysis of differentially expressed transcription factor family
| [1] | Xu YS, Yu D, Chen J, et al. A review of rice male sterility types and their sterility mechanisms[J]. Heliyon, 2023. |
| [2] | 李翔, 李涛, 宫超, 等. 番茄雄性不育研究现状与展望[J]. 广东农业科学, 2023, 50(02): 49-58. |
| Li X, Li T, Gong C, et al. Current situation and prospect of research on male sterility of tomato[J]. Guangdong Agricultural Sciences, 2023, 50(02): 49-58. | |
| [3] | Scott RJ, Spielman M, Dickinson HG. Stamen structure and function[J]. The Plant Cell, 2004, 16(suppl_1): S46-S60. |
| [4] | Kaul MLH. Male sterility in higher plants[M]. Springer Science & Business Media, 2012. |
| [5] |
Chen L, Liu YG. Male sterility and fertility restoration in crops[J]. Annual review of plant biology, 2014, 65: 579-606.
doi: 10.1146/annurev-arplant-050213-040119 pmid: 24313845 |
| [6] | De Storme N, Geelen D. The impact of environmental stress on male reproductive development in plants: biological processes and molecular mechanisms[J]. Plant Cell Environ, 2014, 37(1): 1-18. |
| [7] |
Dukowic-Schulze S, van der Linde K. Oxygen, secreted proteins and small RNAs: mobile elements that govern anther development[J]. Plant Reproduction, 2021, 34(1): 1-19.
doi: 10.1007/s00497-020-00401-0 pmid: 33492519 |
| [8] | 吴智明, 胡开林, 符积钦, 等. 辣椒胞质雄性不育与花蕾内源激素含量的关系[J]. 华南农业大学学报, 2010, 31(2): 1-4. |
| Wu ZM, Hu KL, Fu JQ, et al. Relationships between cytoplasmic male sterility and endogenous hormone content of pepper bud[J]. Journal of South China Agricultural University, 2010, 31(2): 1-4. | |
| [9] | Liu H, Xie WF, Zhang L, et al. Auxin biosynthesis by the YUCCA6 flavin monooxygenase gene in woodland strawberry[J]. Journal of Integrative Plant Biology, 2014, 56(4): 350-363. |
| [10] | Zhang ZB, Zhu J, Gao JF, et al. Transcription factor AtMYB103 is required for anther development by regulating tapetum development, callose dissolution and exine formation in Arabidopsis[J]. The Plant Journal, 2007, 52(3): 528-538. |
| [11] | Thorstensen T, Grini PE, Mercy IS, et al. The Arabidopsis SET-domain protein ASHR3 is involved in stamen development and interacts with the bHLH transcription factor ABORTED MICROSPORES(AMS)[J]. Plant Molecular Biology, 2008, 66(1): 47-59. |
| [12] | Xiong SX, Lu JY, Lou Y, et al. The transcription factors MS 188 and AMS form a complex to activate the expression of CYP703A2 for sporopollenin biosynthesis in Arabidopsis thaliana[J]. The Plant Journal, 2016, 88(6): 936-946. |
| [13] | 刘济铭, 孙操稳, 何秋阳, 等. 国内外无患子属种质资源研究进展[J]. 世界林业研究, 2017, 30(6): 12-18. |
| Liu JM, Sun CW, He QY, et al. Research progress in Sapindus L. germplasm resources[J]. World Forestry Research, 2017, 30(6): 12-18. | |
| [14] | 邵文豪, 刁松锋, 董汝湘, 等. 无患子种实形态及经济性状的地理变异[J]. 林业科学研究, 2013, 26(5): 603-608. |
| Shao WH, Diao SF, Dong RX, et al. Study on geographic variation of morphology and economic character of fruit and seed of Sapindus mukorossi[J]. Forest Research, 2013, 26(5): 603-608. | |
| [15] | 赵国春. 无患子花发育及性别分化机理研究[D]. 北京: 北京林业大学, 2023. |
| Zhao GC. Study on the mechanism of flower development and sex differentiation of Sapindus mukorossi[D]. Beijing: Beijing Forestry University, 2023. | |
| [16] | Xu YY, Zhao GC, Ji XQ, et al. Metabolome and transcriptome analysis reveals the transcriptional regulatory mechanism of triterpenoid saponin biosynthesis in soapberry(Sapindus mukorossi Gaertn.)[J]. Journal of Agricultural and Food Chemistry, 2022, 70(23): 7095-7109. |
| [17] |
徐圆圆, 赵国春, 郝颖颖, 等. 无患子RT-qPCR内参基因的筛选与验证[J]. 生物技术通报, 2022, 38(10): 80-89.
doi: 10.13560/j.cnki.biotech.bull.1985.2021-1616 |
| Xu YY, Zhao GC, Hao YY, et al. Reference genes selection and validation for RT-qPCR in Sapindus mukorossi[J]. Biotechnology Bulletin, 2022, 38(10): 80-89. | |
| [18] |
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2- ΔΔCT method[J]. Methods, 2001, 25(4): 402-408.
doi: 10.1006/meth.2001.1262 pmid: 11846609 |
| [19] |
Jiang J, Zhang Z, Cao J. Pollen wall development: the associated enzymes and metabolic pathways[J]. Plant Biology, 2013, 15(2): 249-263.
doi: 10.1111/j.1438-8677.2012.00706.x pmid: 23252839 |
| [20] | Vu KV, Nguyen NT, Jeong CY, et al. Systematic deletion of the ER lectin chaperone genes reveals their roles in vegetative growth and male gametophyte development in Arabidopsis[J]. The Plant Journal, 2017, 89(5): 972-983. |
| [21] |
Yang K, Zhou XJ, Wang YY, et al. Carbohydrate metabolism and gene regulation during anther development in an androdioecious tree, Tapiscia sinensis[J]. Annals of Botany, 2017, 120(6): 967-977.
doi: 10.1093/aob/mcx094 pmid: 28961748 |
| [22] | Wang R, Dobritsa AA. Exine and aperture patterns on the pollen surface: their formation and roles in plant reproduction[J]. Annual Plant Reviews Online, 2018: 589-628. |
| [23] | Mondol PC, Xu D, Duan L, et al. Defective pollen wall 3(DPW3), a novel alpha integrin-like protein, is required for pollen wall formation in rice[J]. New Phytologist, 2020, 225(2): 807-822. |
| [24] | Li FS, Phyo P, Jacobowitz J, et al. The molecular structure of plant sporopollenin[J]. Nature plants, 2019, 5(1): 41-46. |
| [25] | Tidy AC, Ferjentsikova I, Vizcay-Barrena G, et al. Sporophytic control of pollen meiotic progression is mediated by tapetum expression of ABORTED MICROSPORES[J]. Journal of Experimental Botany, 2022, 73(16): 5543-5558. |
| [26] |
Flores-Tornero M, Anoman AD, Rosa-Téllez S, et al. Lack of phosphoserine phosphatase activity alters pollen and tapetum development in Arabidopsis thaliana[J]. Plant Science, 2015, 235: 81-88.
doi: 10.1016/j.plantsci.2015.03.001 pmid: 25900568 |
| [27] | Chen W, Yu XH, Zhang K, et al. Male Sterile2 encodes a plastid-localized fatty acyl carrier protein reductase required for pollen exine development in Arabidopsis[J]. Plant Physiology, 2011, 157(2): 842-853. |
| [28] | Aarts MG, Keijzer CJ, Stiekema WJ, et al. Molecular characterization of the CER1 gene of arabidopsis involved in epicuticular wax biosynthesis and pollen fertility[J]. The Plant Cell, 1995, 7(12): 2115-2127. |
| [29] | Suzuki T, Narciso JO, Zeng W, et al. KNS4/UPEX1: a type II arabinogalactan β-(1, 3)-galactosyltransferase required for pollen exine development[J]. Plant Physiology, 2017, 173(1): 183-205. |
| [30] | Li JJ, Han SH, Ding XL, et al. Comparative transcriptome analysis between the cytoplasmic male sterile line NJCMS1A and its maintainer NJCMS1B in soybean(Glycine max(L.)Merr.)[J]. PLoS One, 2015, 10(5): e0126771. |
| [31] | Hua MY, Yin WZ, Fernández Gómez J, et al. Barley TAPETAL DEVELOPMENT and FUNCTION1(HvTDF1)gene reveals conserved and unique roles in controlling anther tapetum development in dicot and monocot plants[J]. New Phytologist, 2023, 240(1): 173-190. |
| [32] | Li H, Zhang DB. Biosynthesis of anther cuticle and pollen exine in rice[J]. Plant signaling & behavior, 2010, 5(9): 1121-1123. |
| [33] | Zhao YN, Luo LL, Xu JS, et al. Malate transported from chloroplast to mitochondrion triggers production of ROS and PCD in Arabidopsis thaliana[J]. Cell Research, 2018, 28(4): 448-461. |
| [34] | Liu J, Xia C, Dong HX, et al. Wheat male-sterile 2 reduces ROS levels to inhibit anther development by deactivating ROS modulator 1[J]. Molecular Plant, 2022, 15(9): 1428-1439. |
| [35] | Zhang DD, Liu D, Lv XM, et al. The cysteine protease CEP1, a key executor involved in tapetal programmed cell death, regulates pollen development in Arabidopsis[J]. The Plant Cell, 2014, 26(7): 2939-2961. |
| [36] |
De Veylder L, Beeckman T, Inzé D. The ins and outs of the plant cell cycle[J]. Nature Reviews Molecular Cell Biology, 2007, 8 (8): 655-665.
doi: 10.1038/nrm2227 pmid: 17643126 |
| [37] | Zhang TB, Yuan SH, Liu ZH, et al. Comparative transcriptome analysis reveals hormone signal transduction and sucrose metabolism related genes involved in the regulation of anther dehiscence in photo-thermo-sensitive genic male sterile wheat[J]. Biomolecules, 2022, 12(8): 1149 |
| [38] | He MT, Wang XX, Bu YN, et al. Gibberellin confers to the expression of TaGA-6D and negatively regulates the fertility of wheat with Aegilops juvenalis cytoplasm[J]. Plant Science, 2023: 111771. |
| [39] | Park JH, Halitschke R, Kim HB, et al. A knock-out mutation in allene oxide synthase results in male sterility and defective wound signal transduction in Arabidopsis due to a block in jasmonic acid biosynthesis[J]. The Plant Journal, 2002, 31(1): 1-12. |
| [40] | Li Z, Luo X, Ou Y, et al. JASMONATE-ZIM DOMAIN proteins engage polycomb chromatin modifiers to modulate jasmonate signaling in Arabidopsis[J]. Molecular Plant, 2021, 14(5): 732-747. |
| [41] | Fang YX, Guo DS, Wang Y, et al. Rice transcriptional repressor OsTIE1 controls anther dehiscence and male sterility by regulating JA biosynthesis[J]. The Plant Cell, 2024: koae028. |
| [1] | 高萌萌, 赵天宇, 焦馨悦, 林春晶, 关哲允, 丁孝羊, 孙妍妍, 张春宝. 大豆细胞质雄性不育系及其恢复系的比较转录组分析[J]. 生物技术通报, 2024, 40(7): 137-149. |
| [2] | 秦健, 李振月, 何浪, 李俊玲, 张昊, 杜荣. 肌源性细胞分化的单细胞转录谱变化及细胞间通讯分析[J]. 生物技术通报, 2024, 40(6): 330-342. |
| [3] | 白志元, 徐菲, 杨午, 王明贵, 杨玉花, 张海平, 张瑞军. 大豆细胞质雄性不育弱恢复型杂种F1育性转变的转录组分析[J]. 生物技术通报, 2024, 40(6): 134-142. |
| [4] | 吴迪, 游小凤, 郑亦铮, 林楠, 张燕燕, 魏艺聪. 草珊瑚中类胡萝卜素合成的内源激素调控机制分析[J]. 生物技术通报, 2024, 40(5): 203-214. |
| [5] | 张震, 李清, 徐菁, 陈凯园, 张春芝, 祝光涛. 马铃薯线粒体靶向表达载体的构建与应用[J]. 生物技术通报, 2024, 40(5): 66-73. |
| [6] | 郭纯, 宋桂梅, 闫艳, 邸鹏, 王英平. 西洋参bZIP基因家族全基因组鉴定和表达分析[J]. 生物技术通报, 2024, 40(4): 167-178. |
| [7] | 钟匀, 林春, 刘正杰, 董陈文华, 毛自朝, 李兴玉. 芦笋皂苷合成相关糖基转移酶基因克隆及原核表达分析[J]. 生物技术通报, 2024, 40(4): 255-263. |
| [8] | 杨淇, 魏子迪, 宋娟, 童堃, 杨柳, 王佳涵, 刘海燕, 栾维江, 马轩. 水稻组蛋白H1三突变体的创建和转录组学分析[J]. 生物技术通报, 2024, 40(4): 85-96. |
| [9] | 谢倩, 江来, 贺进, 刘玲玲, 丁明月, 陈清西. 不同鲜食品质橄榄果实转录组测序及酚类代谢途径相关调控基因挖掘[J]. 生物技术通报, 2024, 40(3): 215-228. |
| [10] | 吴圳, 张明英, 闫锋, 李依民, 高静, 颜永刚, 张岗. 掌叶大黄(Rheum palmatum L.)WRKY基因家族鉴定与分析[J]. 生物技术通报, 2024, 40(1): 250-261. |
| [11] | 林红妍, 郭晓蕊, 刘迪, 李慧, 陆海. 转录组分析转录因子AtbHLH68调控细胞壁发育的分子机制[J]. 生物技术通报, 2023, 39(9): 105-116. |
| [12] | 娄慧, 朱金成, 杨洋, 张薇. 抗、感品种棉花根系分泌物对尖孢镰刀菌生长及基因表达的影响[J]. 生物技术通报, 2023, 39(9): 156-167. |
| [13] | 苗永美, 苗翠苹, 于庆才. 枯草芽孢杆菌BBs-27发酵液性质及脂肽对黄色镰刀菌的抑菌作用[J]. 生物技术通报, 2023, 39(9): 255-267. |
| [14] | 付钰, 贾瑞瑞, 何荷, 王良桂, 杨秀莲. 两种砧木楸树嫁接苗生长差异及转录组比较分析[J]. 生物技术通报, 2023, 39(8): 251-261. |
| [15] | 赵金玲, 安磊, 任晓亮. 单细胞转录组测序技术及其在秀丽隐杆线虫中的应用[J]. 生物技术通报, 2023, 39(6): 158-170. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||