







生物技术通报 ›› 2026, Vol. 42 ›› Issue (2): 250-266.doi: 10.13560/j.cnki.biotech.bull.1985.2025-0481
庄娜( ), 贺红利, 张兴政()
), 贺红利, 张兴政()
收稿日期:2025-05-13
出版日期:2026-02-26
发布日期:2026-03-17
通讯作者:
张兴政,男,博士,副教授,研究方向 :植物分子遗传学;E-mail: zxz20051986@163.com作者简介:庄娜,女,研究方向 :植物分子遗传学;E-mail: m15589101772@163.com
基金资助:
ZHUANG Na(), HE Hong-li, ZHANG Xing-zheng()
Received:2025-05-13
Published:2026-02-26
Online:2026-03-17
摘要:
目的 SMXLs(SUPPRESSOR of MAX2 1-LIKE)是重要植物激素独角金内酯信号转导通路中的抑制因子及烟素(karrikins, KARs)信号通路的主要成员。在全基因组水平鉴定蒺藜苜蓿SMXL基因家族成员并分析其在非生物胁迫下的表达模式变化,为豆科植物相关基因家族的研究提供理论支持,为未来深入研究MtSMXL基因的功能奠定基础。 方法 运用生物信息学方法,基于已报道植物SMXL家族编码氨基酸序列,对蒺藜苜蓿SMXL家族成员进行全基因组鉴定,利用TBtools等工具对MtSMXL成员进行进化和功能分析,同时利用转录组数据进行成员不同组织内表达分析及钾、氮、磷、硫营养缺乏胁迫处理和在冷害、干旱、高盐胁迫下的表达量分析。 结果 依据进化分析,共筛选鉴定出17个MtSMXL成员。参考基因结构和保守结构域将它们分为G1-G3三个组群;MtSMXLs上游启动子区域存在多种参与响应植物激素和非生物胁迫的顺式作用元件,各成员中类型和数量有一定差异;共线性分析表明,豆科植物WGD事件对MtSMXL基因家族的扩张有重要作用;家族成员在不同组织部位及响应各种胁迫的过程中其表达模式均存在明显差异,通过分析选出参与不同胁迫响应功能候选基因。 结论 蒺藜苜蓿SMXL基因不同组群在进化中结构和功能差异较大,成员及不同胁迫下响应模式不同,其中MtSMXL2/7/11可用于K/N/S/P元素缺乏胁迫;而MtSMXL7/8/13/14/16可用于干旱、冷、高盐胁迫响应通路研究。
庄娜, 贺红利, 张兴政. 蒺藜苜蓿SMXL基因家族全基因组鉴定及表达分析[J]. 生物技术通报, 2026, 42(2): 250-266.
ZHUANG Na, HE Hong-li, ZHANG Xing-zheng. Genome-wide Identification and Expression Analysis of the SMXL Gene Family in Medicago truncatula[J]. Biotechnology Bulletin, 2026, 42(2): 250-266.
| 基因名称 Gene name | 基因ID Gene ID | 染色体定位 Location | 基因链 Strand | 外显子数 Exon | 编码区长度 CDS (bp) |
|---|---|---|---|---|---|
| MtSMXL1 | Medtr1g057150 | chr1:25117416..25121709 | + | 3 | 2 427 |
| MtSMXL2 | Medtr1g090640 | chr1:40585935..40589246 | - | 3 | 2 550 |
| MtSMXL3 | Medtr2g038200 | chr2:16610767..16615642 | + | 3 | 3 030 |
| MtSMXL4 | Medtr3g461000 | chr3:24065055..24068423 | + | 8 | 2 449 |
| MtSMXL5 | Medtr3g464150 | chr3:25772408..25778862 | + | 3 | 3 243 |
| MtSMXL6 | Medtr3g070850 | chr3:31780989..31785558 | + | 3 | 3 246 |
| MtSMXL7 | Medtr3g098310 | chr3:44871095..44878208 | - | 13 | 2 859 |
| MtSMXL8 | Medtr3g498725 | chr3:45109729..45116929 | + | 11 | 2 769 |
| MtSMXL9 | Medtr3g102830 | chr3:47377876..47383774 | + | 10 | 2 784 |
| MtSMXL10 | Medtr3g106160 | chr3:49006958..49015432 | - | 10 | 2 943 |
| MtSMXL11 | Medtr4g109450 | chr4:45465442..45469308 | + | 5 | 2 739 |
| MtSMXL12 | Medtr4g127290 | chr4:52803794..52807296 | - | 3 | 2 463 |
| MtSMXL13 | Medtr4g129230 | chr4:53817793..53821564 | + | 3 | 3 126 |
| MtSMXL14 | Medtr5g071060 | chr5:30105997..30111708 | + | 3 | 3 279 |
| MtSMXL15 | Medtr6g013660 | chr6:4395062..4403563 | - | 10 | 2 925 |
| MtSMXL16 | Medtr7g012820 | chr7:3513549..3521223 | - | 10 | 2 937 |
| MtSMXL17 | Medtr8g100040 | chr8:40241178..40246683 | + | 10 | 2 781 |
表1 MtSMXL基因基础信息
Table 1 Basic information of MtSMXL genes
| 基因名称 Gene name | 基因ID Gene ID | 染色体定位 Location | 基因链 Strand | 外显子数 Exon | 编码区长度 CDS (bp) |
|---|---|---|---|---|---|
| MtSMXL1 | Medtr1g057150 | chr1:25117416..25121709 | + | 3 | 2 427 |
| MtSMXL2 | Medtr1g090640 | chr1:40585935..40589246 | - | 3 | 2 550 |
| MtSMXL3 | Medtr2g038200 | chr2:16610767..16615642 | + | 3 | 3 030 |
| MtSMXL4 | Medtr3g461000 | chr3:24065055..24068423 | + | 8 | 2 449 |
| MtSMXL5 | Medtr3g464150 | chr3:25772408..25778862 | + | 3 | 3 243 |
| MtSMXL6 | Medtr3g070850 | chr3:31780989..31785558 | + | 3 | 3 246 |
| MtSMXL7 | Medtr3g098310 | chr3:44871095..44878208 | - | 13 | 2 859 |
| MtSMXL8 | Medtr3g498725 | chr3:45109729..45116929 | + | 11 | 2 769 |
| MtSMXL9 | Medtr3g102830 | chr3:47377876..47383774 | + | 10 | 2 784 |
| MtSMXL10 | Medtr3g106160 | chr3:49006958..49015432 | - | 10 | 2 943 |
| MtSMXL11 | Medtr4g109450 | chr4:45465442..45469308 | + | 5 | 2 739 |
| MtSMXL12 | Medtr4g127290 | chr4:52803794..52807296 | - | 3 | 2 463 |
| MtSMXL13 | Medtr4g129230 | chr4:53817793..53821564 | + | 3 | 3 126 |
| MtSMXL14 | Medtr5g071060 | chr5:30105997..30111708 | + | 3 | 3 279 |
| MtSMXL15 | Medtr6g013660 | chr6:4395062..4403563 | - | 10 | 2 925 |
| MtSMXL16 | Medtr7g012820 | chr7:3513549..3521223 | - | 10 | 2 937 |
| MtSMXL17 | Medtr8g100040 | chr8:40241178..40246683 | + | 10 | 2 781 |
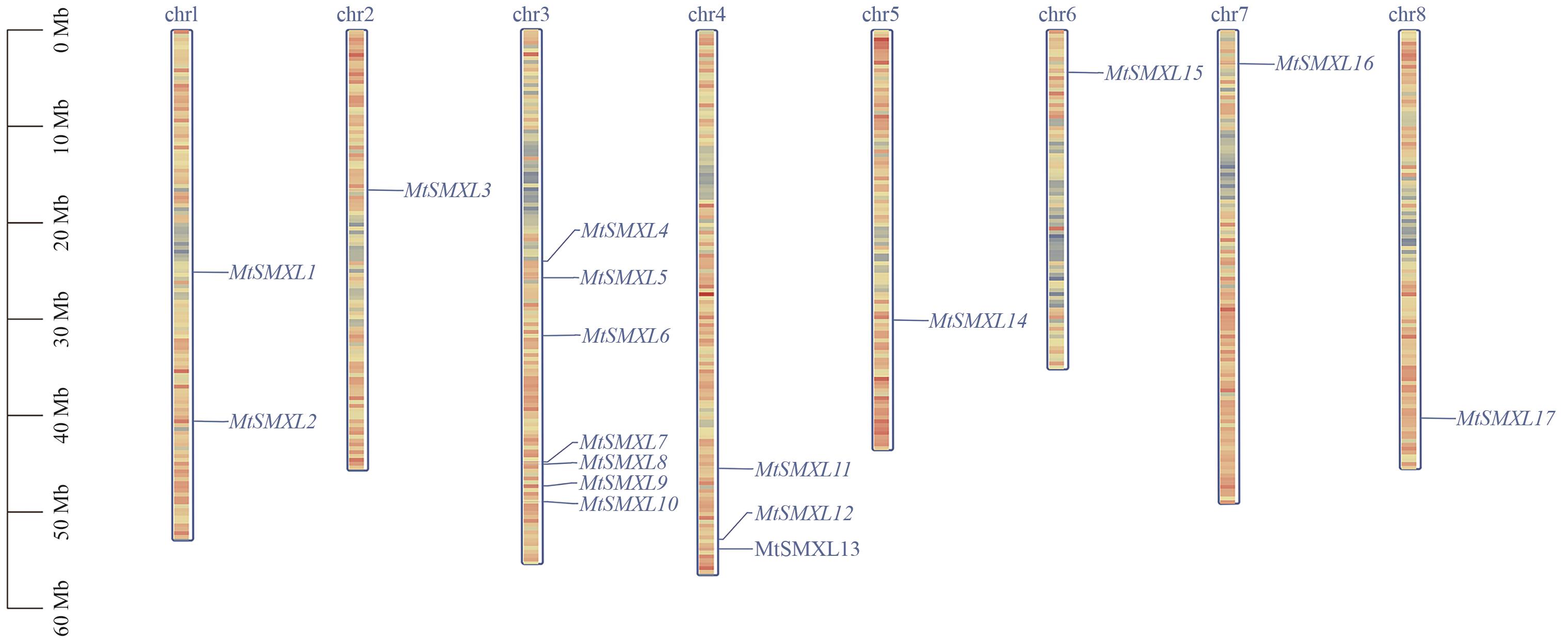
图1 蒺藜苜蓿SMXL基因家族成员染色体分布红色代表高基因密度,蓝色代表低基因密度
Fig. 1 Chromosome distribution of SMXL gene family in M. truncatulaRed indicates high gene density, and blue indicates low gene density
基因名称 Gene name | 氨基酸数 Peptide (aa) | 分子质量 Mass of molecule (kD) | 等电点 Theoretical pI | 不稳定系数 Instability index | 脂肪指数 Aliphatic index | 平均亲水指数 GRAVY | 亚细胞定位 Subcellular localization |
|---|---|---|---|---|---|---|---|
| MtSMXL1 | 808 | 90.50 | 5.74 | 46.88 | 82.86 | -0.502 | 叶绿体 |
| MtSMXL2 | 849 | 95.17 | 6.03 | 54.29 | 82.99 | -0.362 | 叶绿体 |
| MtSMXL3 | 1 009 | 113.46 | 6.41 | 51.71 | 78.12 | -0.509 | 细胞核 |
| MtSMXL4 | 810 | 90.80 | 7.33 | 37.38 | 97.32 | -0.233 | 细胞质 |
| MtSMXL5 | 1 080 | 120.931 | 6.27 | 50.68 | 85.24 | -0.307 | 细胞核 |
| MtSMXL6 | 1 081 | 119.74 | 6.34 | 50.45 | 84.38 | -0.356 | 细胞核 |
| MtSMXL7 | 963 | 107.09 | 7.04 | 47.16 | 89.41 | -0.287 | 叶绿体 |
| MtSMXL8 | 922 | 102.71 | 6.35 | 40.68 | 95.40 | -0.341 | 叶绿体 |
| MtSMXL9 | 927 | 103.07 | 6.41 | 40.14 | 93.50 | -0.360 | 叶绿体 |
| MtSMXL10 | 980 | 109.89 | 6.53 | 38.67 | 92.85 | -0.385 | 线粒体 |
| MtSMXL11 | 912 | 101.35 | 5.85 | 36.33 | 96.23 | -0.402 | 细胞质 |
| MtSMXL12 | 820 | 92.33 | 8.18 | 58.46 | 77.52 | -0.524 | 叶绿体 |
| MtSMXL13 | 1 027 | 112.90 | 6.34 | 46.61 | 83.50 | -0.471 | 细胞质 |
| MtSMXL14 | 1 092 | 120.61 | 6.05 | 43.61 | 81.12 | -0.254 | 细胞核 |
| MtSMXL15 | 974 | 109.96 | 6.65 | 42.89 | 89.30 | -0.474 | 叶绿体 |
| MtSMXL16 | 978 | 110.28 | 6.26 | 37.80 | 90.14 | -0.455 | 线粒体 |
| MtSMXL17 | 926 | 102.78 | 6.44 | 40.51 | 94.04 | -0.357 | 叶绿体 |
表2 MtSMXL蛋白理化性质预测
Table 2 Prediction of physical and chemical properties of MtSMXLproteins
基因名称 Gene name | 氨基酸数 Peptide (aa) | 分子质量 Mass of molecule (kD) | 等电点 Theoretical pI | 不稳定系数 Instability index | 脂肪指数 Aliphatic index | 平均亲水指数 GRAVY | 亚细胞定位 Subcellular localization |
|---|---|---|---|---|---|---|---|
| MtSMXL1 | 808 | 90.50 | 5.74 | 46.88 | 82.86 | -0.502 | 叶绿体 |
| MtSMXL2 | 849 | 95.17 | 6.03 | 54.29 | 82.99 | -0.362 | 叶绿体 |
| MtSMXL3 | 1 009 | 113.46 | 6.41 | 51.71 | 78.12 | -0.509 | 细胞核 |
| MtSMXL4 | 810 | 90.80 | 7.33 | 37.38 | 97.32 | -0.233 | 细胞质 |
| MtSMXL5 | 1 080 | 120.931 | 6.27 | 50.68 | 85.24 | -0.307 | 细胞核 |
| MtSMXL6 | 1 081 | 119.74 | 6.34 | 50.45 | 84.38 | -0.356 | 细胞核 |
| MtSMXL7 | 963 | 107.09 | 7.04 | 47.16 | 89.41 | -0.287 | 叶绿体 |
| MtSMXL8 | 922 | 102.71 | 6.35 | 40.68 | 95.40 | -0.341 | 叶绿体 |
| MtSMXL9 | 927 | 103.07 | 6.41 | 40.14 | 93.50 | -0.360 | 叶绿体 |
| MtSMXL10 | 980 | 109.89 | 6.53 | 38.67 | 92.85 | -0.385 | 线粒体 |
| MtSMXL11 | 912 | 101.35 | 5.85 | 36.33 | 96.23 | -0.402 | 细胞质 |
| MtSMXL12 | 820 | 92.33 | 8.18 | 58.46 | 77.52 | -0.524 | 叶绿体 |
| MtSMXL13 | 1 027 | 112.90 | 6.34 | 46.61 | 83.50 | -0.471 | 细胞质 |
| MtSMXL14 | 1 092 | 120.61 | 6.05 | 43.61 | 81.12 | -0.254 | 细胞核 |
| MtSMXL15 | 974 | 109.96 | 6.65 | 42.89 | 89.30 | -0.474 | 叶绿体 |
| MtSMXL16 | 978 | 110.28 | 6.26 | 37.80 | 90.14 | -0.455 | 线粒体 |
| MtSMXL17 | 926 | 102.78 | 6.44 | 40.51 | 94.04 | -0.357 | 叶绿体 |
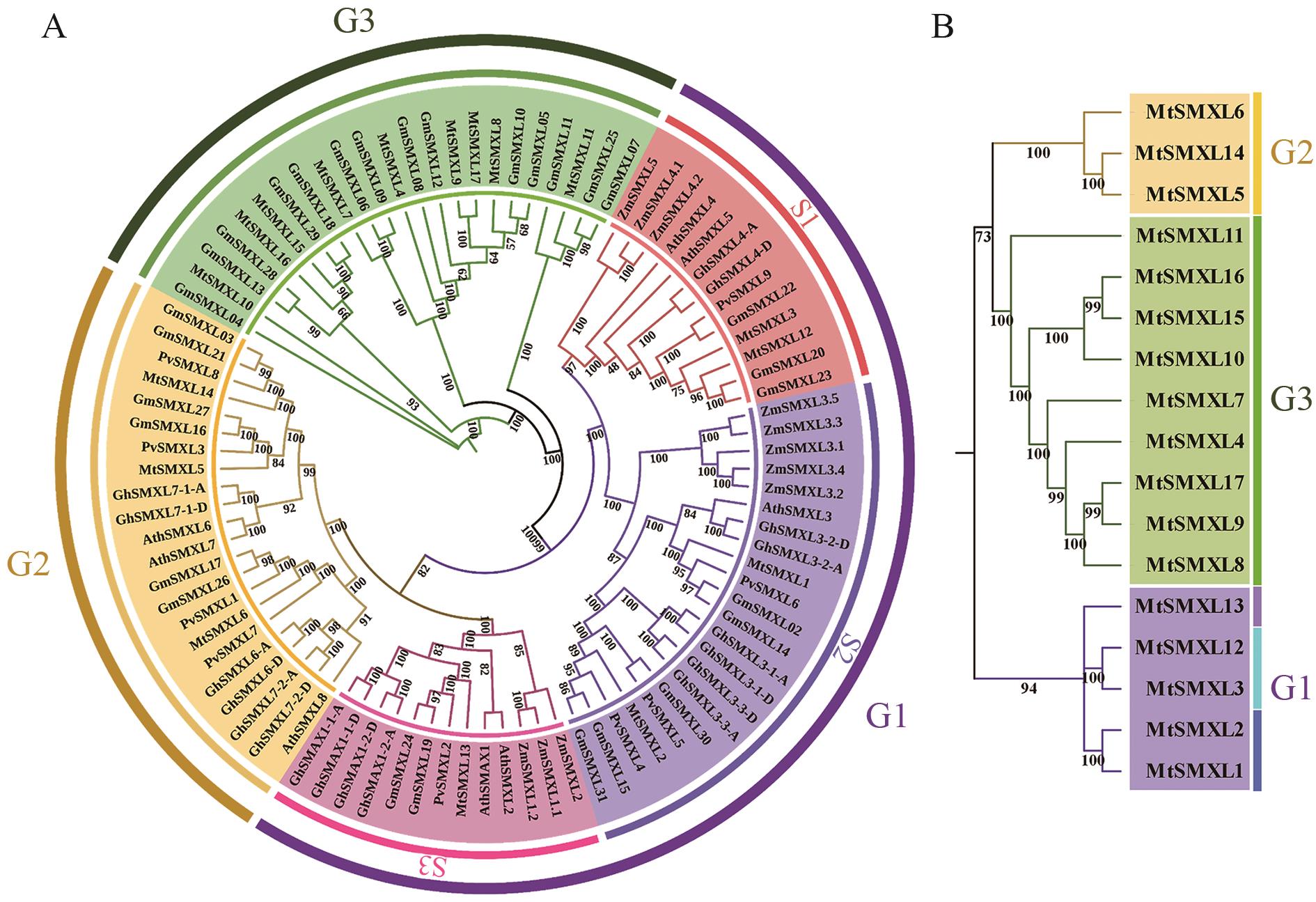
图2 SMXL 基因家族系统发育树A:种间系统发育树;B:种内系统发育树
Fig. 2 Phylogenetic tree of SMXL gene familyA: Interspecific phylogenetic tree. B: Intraspecific phylogenetic tree
蛋白质 Protein | α螺旋 Alpha helix | 延伸链 Extended strand | β转角 β-turn | 无规则卷曲 Random coil | ||||
|---|---|---|---|---|---|---|---|---|
长度 Length (aa) | 所占比例 Proportion (%) | 长度 Length (aa) | 所占比例 Proportion (%) | 长度 Length (aa) | 所占比例 Proportion (%) | 长度 Length (aa) | 所占比例 Proportion (%) | |
| MtSMXL1 | 382 | 47.28 | 68 | 8.42 | 21 | 2.60 | 358 | 44.31 |
| MtSMXL2 | 366 | 43.11 | 53 | 6.24 | 13 | 1.53 | 430 | 50.65 |
| MtSMXL3 | 440 | 43.61 | 77 | 7.63 | 17 | 1.68 | 492 | 48.76 |
| MtSMXL4 | 417 | 51.48 | 87 | 10.74 | 51 | 6.30 | 306 | 37.78 |
| MtSMXL5 | 443 | 41.02 | 88 | 8.15 | 14 | 1.30 | 549 | 50.83 |
| MtSMXL6 | 406 | 37.56 | 99 | 9.16 | 19 | 1.76 | 576 | 53.28 |
| MtSMXL7 | 506 | 52.54 | 86 | 8.93 | 51 | 5.30 | 371 | 38.53 |
| MtSMXL8 | 496 | 53.80 | 87 | 9.44 | 51 | 5.53 | 339 | 36.77 |
| MtSMXL9 | 523 | 56.42 | 85 | 9.17 | 50 | 5.39 | 319 | 34.41 |
| MtSMXL10 | 563 | 57.45 | 82 | 8.37 | 44 | 4.49 | 335 | 34.18 |
| MtSMXL11 | 550 | 60.31 | 84 | 9.21 | 45 | 4.93 | 278 | 30.48 |
| MtSMXL12 | 363 | 44.27 | 53 | 6.46 | 7 | 0.85 | 404 | 49.27 |
| MtSMXL13 | 512 | 49.85 | 70 | 6.82 | 20 | 1.95 | 445 | 43.33 |
| MtSMXL14 | 471 | 43.13 | 84 | 7.69 | 16 | 1.47 | 537 | 49.18 |
| MtSMXL15 | 554 | 56.88 | 84 | 8.62 | 46 | 4.72 | 336 | 34.50 |
| MtSMXL16 | 545 | 55.73 | 86 | 8.79 | 45 | 4.60 | 347 | 35.48 |
| MtSMXL17 | 507 | 54.75 | 92 | 9.94 | 49 | 5.29 | 327 | 35.31 |
表3 MtSMXLs蛋白质二级结构预测
Table 3 Secondary structure prediction of the MtSMXLs protein
蛋白质 Protein | α螺旋 Alpha helix | 延伸链 Extended strand | β转角 β-turn | 无规则卷曲 Random coil | ||||
|---|---|---|---|---|---|---|---|---|
长度 Length (aa) | 所占比例 Proportion (%) | 长度 Length (aa) | 所占比例 Proportion (%) | 长度 Length (aa) | 所占比例 Proportion (%) | 长度 Length (aa) | 所占比例 Proportion (%) | |
| MtSMXL1 | 382 | 47.28 | 68 | 8.42 | 21 | 2.60 | 358 | 44.31 |
| MtSMXL2 | 366 | 43.11 | 53 | 6.24 | 13 | 1.53 | 430 | 50.65 |
| MtSMXL3 | 440 | 43.61 | 77 | 7.63 | 17 | 1.68 | 492 | 48.76 |
| MtSMXL4 | 417 | 51.48 | 87 | 10.74 | 51 | 6.30 | 306 | 37.78 |
| MtSMXL5 | 443 | 41.02 | 88 | 8.15 | 14 | 1.30 | 549 | 50.83 |
| MtSMXL6 | 406 | 37.56 | 99 | 9.16 | 19 | 1.76 | 576 | 53.28 |
| MtSMXL7 | 506 | 52.54 | 86 | 8.93 | 51 | 5.30 | 371 | 38.53 |
| MtSMXL8 | 496 | 53.80 | 87 | 9.44 | 51 | 5.53 | 339 | 36.77 |
| MtSMXL9 | 523 | 56.42 | 85 | 9.17 | 50 | 5.39 | 319 | 34.41 |
| MtSMXL10 | 563 | 57.45 | 82 | 8.37 | 44 | 4.49 | 335 | 34.18 |
| MtSMXL11 | 550 | 60.31 | 84 | 9.21 | 45 | 4.93 | 278 | 30.48 |
| MtSMXL12 | 363 | 44.27 | 53 | 6.46 | 7 | 0.85 | 404 | 49.27 |
| MtSMXL13 | 512 | 49.85 | 70 | 6.82 | 20 | 1.95 | 445 | 43.33 |
| MtSMXL14 | 471 | 43.13 | 84 | 7.69 | 16 | 1.47 | 537 | 49.18 |
| MtSMXL15 | 554 | 56.88 | 84 | 8.62 | 46 | 4.72 | 336 | 34.50 |
| MtSMXL16 | 545 | 55.73 | 86 | 8.79 | 45 | 4.60 | 347 | 35.48 |
| MtSMXL17 | 507 | 54.75 | 92 | 9.94 | 49 | 5.29 | 327 | 35.31 |
| 基序 Motif | Sites | 长度 Length (aa) | 序列 Sequence | Logo |
|---|---|---|---|---|
| 1 | 9 | 50 | MSEYMERHTVSRLIGAPPGYVGYEEGGQLTEAVRRRPYTVVLFDEIEKAH |  |
| 2 | 9 | 50 | GATTLDEYRKHIEKDPALERRFQPVKVPEPSVEDTISILKGLRERYEJHH |  |
| 3 | 9 | 50 | GKLDPVIGRDEZIERVIQILSRRTKNNPVLIGEPGVGKTAIAEGLAQRIV |  |
| 4 | 17 | 41 | NPNRPIASFMFLGPTGVGKTELAKALAEYVFGSEEALIRJD |  |
| 5 | 15 | 35 | LQIJEDGRLTDSQGRTVSFKNTIIIMTSNVGSSVI |  |
| 6 | 17 | 38 | QZELTEEAAKVJKLAVEVARRRGHAQVTPLHIASALLS |  |
| 7 | 9 | 50 | KVRYTDDALVAAAELSDRYISDRFLPDKAIDLIDEAGSKVRLZHASKPEE |  |
| 8 | 7 | 50 | VLKEVTESSGZIILFIDEIHTVIGAGATEGAMDAANJLKPMLARGELRCI |  |
| 9 | 9 | 49 | WTGIPVSKLSQDERERLLKLEDTLHKRVIGQDEAVEAIARAIRRSRVGL |  |
| 10 | 9 | 50 | KDSSYNRIKSLVMEELRQYFRPEFLNRLDEMIVFRPLTKLZVKEIADJML |  |
| 11 | 9 | 50 | VTERARDLVVDEGYBPSYGARPLRRAIQRLVEBELAEKMLRREIKEGDSV |  |
| 12 | 8 | 29 | VKVELEQLIJSILDDPSVSRVMREAGFSS |  |
| 13 | 9 | 24 | PPVSNALMAAJKRAQAHQRRGPIE |  |
| 14 | 17 | 29 | DVPETLHPLQLIALDLGLLVALAKYPGEF |  |
| 15 | 4 | 50 | ARQLGHNYIGSEHLLLGLLREGEGVAARVLENLGADPTNIRTQVIRMVGE |  |
表4 MtSMXLs保守基序序列鉴定
Table 4 Conserved motifs identification of MtSMXLs
| 基序 Motif | Sites | 长度 Length (aa) | 序列 Sequence | Logo |
|---|---|---|---|---|
| 1 | 9 | 50 | MSEYMERHTVSRLIGAPPGYVGYEEGGQLTEAVRRRPYTVVLFDEIEKAH | |
| 2 | 9 | 50 | GATTLDEYRKHIEKDPALERRFQPVKVPEPSVEDTISILKGLRERYEJHH | |
| 3 | 9 | 50 | GKLDPVIGRDEZIERVIQILSRRTKNNPVLIGEPGVGKTAIAEGLAQRIV | |
| 4 | 17 | 41 | NPNRPIASFMFLGPTGVGKTELAKALAEYVFGSEEALIRJD | |
| 5 | 15 | 35 | LQIJEDGRLTDSQGRTVSFKNTIIIMTSNVGSSVI | |
| 6 | 17 | 38 | QZELTEEAAKVJKLAVEVARRRGHAQVTPLHIASALLS | |
| 7 | 9 | 50 | KVRYTDDALVAAAELSDRYISDRFLPDKAIDLIDEAGSKVRLZHASKPEE | |
| 8 | 7 | 50 | VLKEVTESSGZIILFIDEIHTVIGAGATEGAMDAANJLKPMLARGELRCI | |
| 9 | 9 | 49 | WTGIPVSKLSQDERERLLKLEDTLHKRVIGQDEAVEAIARAIRRSRVGL | |
| 10 | 9 | 50 | KDSSYNRIKSLVMEELRQYFRPEFLNRLDEMIVFRPLTKLZVKEIADJML | |
| 11 | 9 | 50 | VTERARDLVVDEGYBPSYGARPLRRAIQRLVEBELAEKMLRREIKEGDSV | |
| 12 | 8 | 29 | VKVELEQLIJSILDDPSVSRVMREAGFSS | |
| 13 | 9 | 24 | PPVSNALMAAJKRAQAHQRRGPIE | |
| 14 | 17 | 29 | DVPETLHPLQLIALDLGLLVALAKYPGEF | |
| 15 | 4 | 50 | ARQLGHNYIGSEHLLLGLLREGEGVAARVLENLGADPTNIRTQVIRMVGE | |
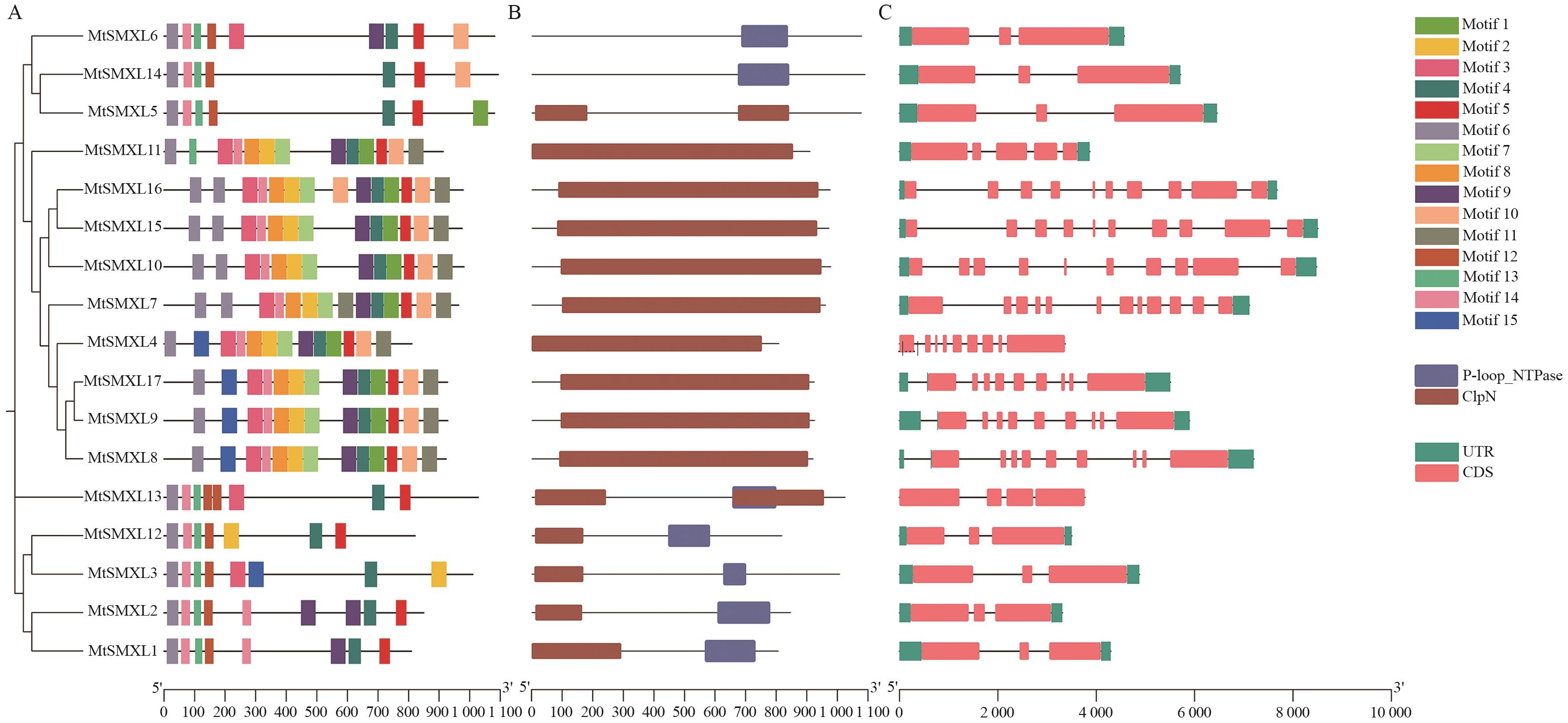
图3 蒺藜苜蓿SMXL成员保守基序、蛋白保守结构域和基因结构A:MtSMXL蛋白的保守基序,不同的方形代表不同的基序;B:MtSMXL蛋白保守结构域,不同的方形代表不同的蛋白质结构域;C:MtSMXLs基因结构
Fig. 3 Conserved motifs, protein conserved domains, and gene structure of MtSMXLs in M. truncatulaA: Conserved motif of MtSMXL protein, different squares indicate different motif. B: MtSMXL protein domain, different squares indicate different protein domains. C: Gene structure of MtSMXL genes
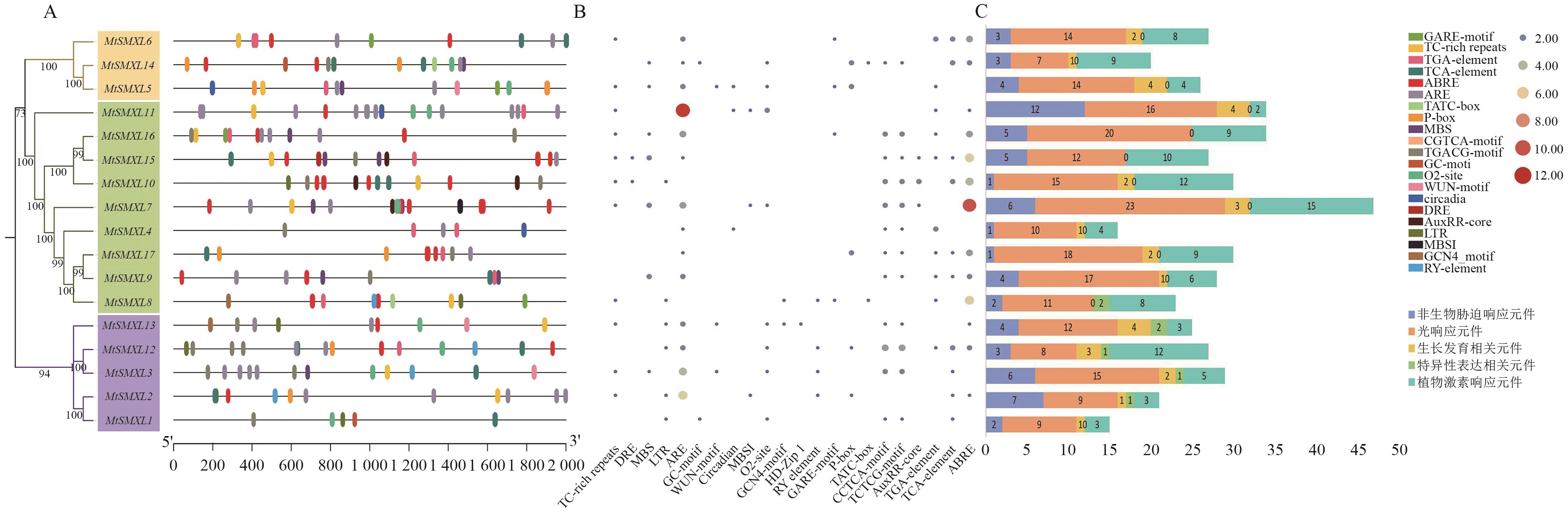
图4 SMXL基因启动子的顺式作用元件A:MtSMXL基因启动子区顺式作用元件分布,不同颜色的椭圆代表不同的元件;B:M启动子区域顺式作用元件数量热图;C:MtSMXL基因启动子区域各类顺式作用元件堆积统计图。由于启动子位置重合和篇幅原因,启动子位置图A和热图B中不展示光响应元件和一些与本研究无关的生长发育相关元件
Fig. 4 Cis-acting elements in the promoter of the SMXL genesA: Distribution of cis-acting element in the promoter region of the MtSMXL gene, with different colored ovals representing different cis-elements. B: Heatmap of the number of cis-acting elements in the promoter region of M . C: Stacked statistical chart of various cis-acting elements in the promoter region of the MtSMXL gene. Due to the overlap of promoter positions and space limitations, light-responsive elements and some growth- and development-related elements irrelevant to this study are not shown in the promoter position map A and the heatmap B
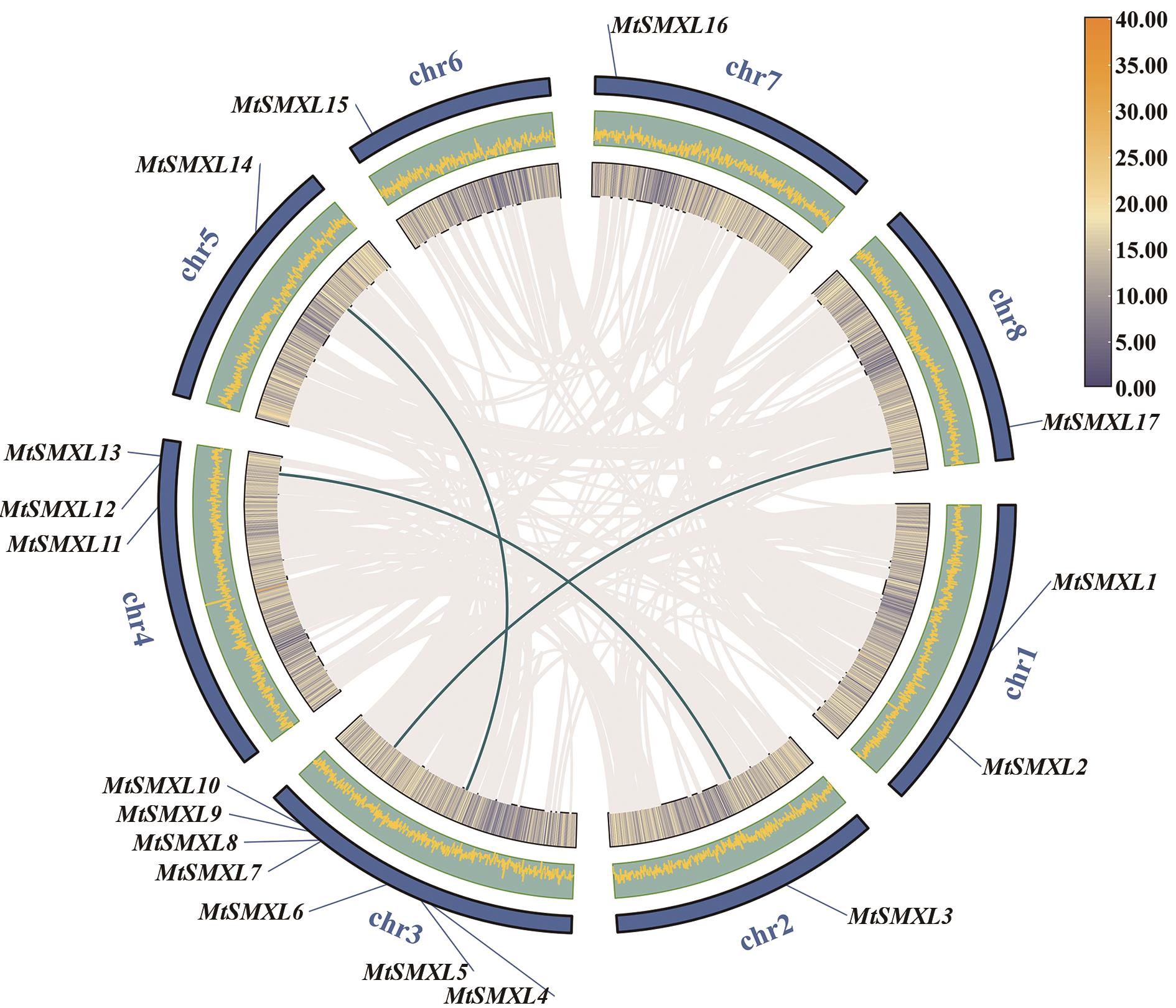
图5 MtSMXL基因物种内共线性图灰色线代表基因组中具有共线性的基因对,绿色线表示具有共线关系的MtSMXL基因对;图中内圈和中圈皆表示蒺藜苜蓿基因组基因密度,最外圈表示染色体;图例表示内圈中基因密度情况,黄色表示高基因密度,蓝色表示低基因密度
Fig. 5 Intraspecific collinearity of the MtSMXL genesThe gray lines indicate collinearity gene pairs in the genome, and the green lines indicate gene pairs ofcollinear MtSMXL. The inner and middle circles indicate the gene density of the M. truncatula genome, and the outermost circle indicates the chromosomes. The legend indicates the gene density in the inner circle, where yellow shows high gene density and blue shows low gene density
| 基因对Gene pairs | Ka | Ks | Ka/Ks | 分离时间Estimated time(Mya) | |
|---|---|---|---|---|---|
| MtSMXL3 | MtSMXL12 | 0.253 | 0.919 | 0.275 | 75.3 |
| MtSMXL5 | MtSMXL14 | 0.255 | 0.721 | 0.354 | 59.08 |
| MtSMXL8 | MtSMXL17 | 0.056 | 0.68 | 0.082 | 55.74 |
表5 MtSMXL基因对分离时间估算
Table 5 Estimated separation time of MtSMXL gene pairs
| 基因对Gene pairs | Ka | Ks | Ka/Ks | 分离时间Estimated time(Mya) | |
|---|---|---|---|---|---|
| MtSMXL3 | MtSMXL12 | 0.253 | 0.919 | 0.275 | 75.3 |
| MtSMXL5 | MtSMXL14 | 0.255 | 0.721 | 0.354 | 59.08 |
| MtSMXL8 | MtSMXL17 | 0.056 | 0.68 | 0.082 | 55.74 |
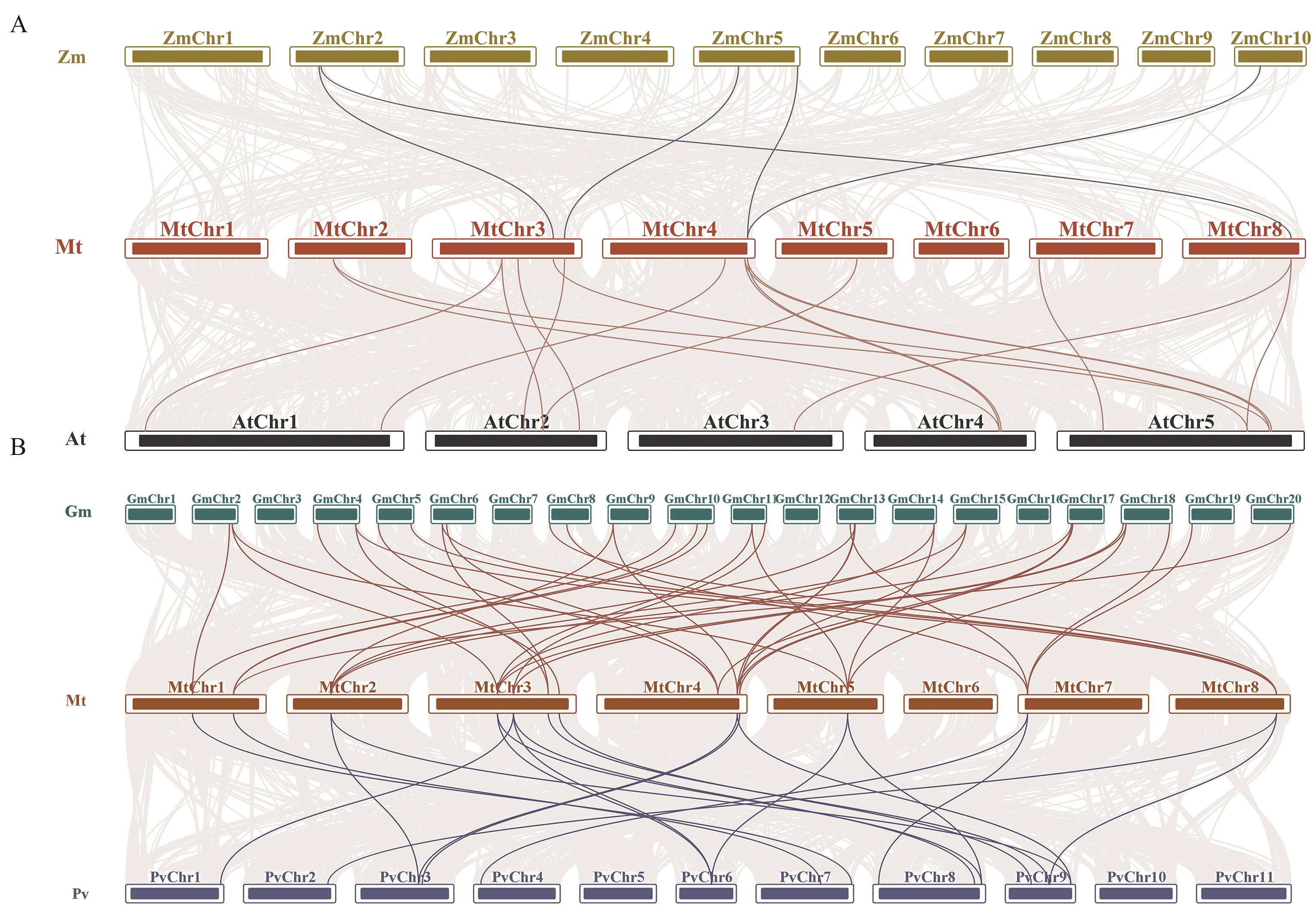
图6 蒺藜苜蓿与其他物种SMXL家族基因的共线性分析A:蒺藜苜蓿与大豆和菜豆的SMXL共线性分析;B:蒺藜苜蓿与玉米和拟南芥的SMXL共线性分析。蒺藜苜蓿和其他物种基因组中的共线由灰线显示,与其他物种之间的共线SMXL基因对用亮色线条突出显示
Fig. 6 Collinearity analysis of SMXL genes between M. truncatula and other speciesA: Collinearity analysis of SMXL family genes in M. truncatula, Glycine max and common bean. B: Collinearity analysis of SMXL family genes in M. truncatula, Z. mays and Arabidopsis. The collinear regions in the genomes of M. truncatula and other species are shown by gray lines, and the collinear SMXL gene pairs with other species are highlighted by bright lines
蒺藜苜蓿 M. truncatula | 拟南芥 A. thaliana | 玉米 Z. mays | 菜豆 P. vulgaris | 大豆 G. max | 蒺藜苜蓿 M. truncatula | 拟南芥 A. thaliana | 玉米 Z. mays | 菜豆 P. vulgaris | 大豆 G. max |
|---|---|---|---|---|---|---|---|---|---|
| MtSMXL1 | ESW17795 | Glyma.02G198000.1 | MtSMXL11 | AT1G74310.1 | Glyma.05G022200.1 | ||||
| Glyma.10G079000.1 | Glyma.06G202200.2 | ||||||||
| MtSMXL2 | ESW16225 | Glyma.10G121542.2 | Glyma.17G077500.1 | ||||||
| Glyma.10G196300.1 | MtSMXL12 | AT4G29920.2 | ESW10759 | Glyma.09G060500.1 | |||||
| Glyma.20G193900.1 | AT5G57130.1 | ESW26697 | Glyma.13G101300.1 | ||||||
| MtSMXL3 | AT4G29920.2 | ESW10759 | Glyma.09G060500.1 | Glyma.15G167000.1 | |||||
| AT5G57130.1 | ESW26697 | Glyma.13G101300.1 | Glyma.17G058400.1 | ||||||
| Glyma.15G167000.1 | MtSMXL13 | AT4G30350.1 | Zm00001eb412800_T002 | ESW26813 | Glyma.13G092700.1 | ||||
| Glyma.17G058400.1 | AT5G57710.1 | Zm00001eb257430_T002 | Glyma.17G067700.1 | ||||||
| MtSMXL4 | MtSMXL14 | AT2G29970.1 | ESW14200 | Glyma.02G226900.6 | |||||
| MtSMXL5 | AT1G07200.2 | ESW14200 | Glyma.02G226900.6 | ESW18766 | Glyma.11G174500.4 | ||||
| AT2G29970.1 | ESW18766 | Glyma.11G174500.4 | Glyma.14G193900.2 | ||||||
| Glyma.14G193900.2 | Glyma.18G062300.1 | ||||||||
| Glyma.18G062300.1 | MtSMXL15 | ||||||||
| MtSMXL6 | AT2G40130.2 | ESW13755 | Glyma.11G230700.1 | MtSMXL16 | AT5G15450.1 | ESW11501 | Glyma.08G242100.1 | ||
| ESW18766 | Glyma.18G026600.3 | ESW23286 | Glyma.13G058600.1 | ||||||
| ESW35546 | Glyma.18G062300.1 | Glyma.18G264400.1 | |||||||
| MtSMXL7 | AT5G51070.1 | Zm00001eb084420_T004 | ESW09825 | Glyma.04G203300.1 | Glyma.19G027900.1 | ||||
| Glyma.06G162200.1 | MtSMXL17 | AT3G48870.3 | Zm00001eb084890_T001 | ESW09858 | Glyma.04G200400.8 | ||||
| MtSMXL8 | AT5G50920.1 | ESW31965 | Glyma.05G201100.4 | ||||||
| MtSMXL9 | Glyma.06G165200.6 | ||||||||
| MtSMXL10 | AT2G25140.1 | Zm00001eb234160_T001 | ESW08954 | Glyma.04G062200.1 | Glyma.08G008400.1 |
表6 MtSMXLs和AtSMXLs、GmSMXLs、PvSMXLs、ZmSMXLs基因的同线性分析
Table 6 Synteny analysis of MdSMXLs and AtSMXLs, GmSMXLs, PvSMXLs, and ZmSMXLs gene
蒺藜苜蓿 M. truncatula | 拟南芥 A. thaliana | 玉米 Z. mays | 菜豆 P. vulgaris | 大豆 G. max | 蒺藜苜蓿 M. truncatula | 拟南芥 A. thaliana | 玉米 Z. mays | 菜豆 P. vulgaris | 大豆 G. max |
|---|---|---|---|---|---|---|---|---|---|
| MtSMXL1 | ESW17795 | Glyma.02G198000.1 | MtSMXL11 | AT1G74310.1 | Glyma.05G022200.1 | ||||
| Glyma.10G079000.1 | Glyma.06G202200.2 | ||||||||
| MtSMXL2 | ESW16225 | Glyma.10G121542.2 | Glyma.17G077500.1 | ||||||
| Glyma.10G196300.1 | MtSMXL12 | AT4G29920.2 | ESW10759 | Glyma.09G060500.1 | |||||
| Glyma.20G193900.1 | AT5G57130.1 | ESW26697 | Glyma.13G101300.1 | ||||||
| MtSMXL3 | AT4G29920.2 | ESW10759 | Glyma.09G060500.1 | Glyma.15G167000.1 | |||||
| AT5G57130.1 | ESW26697 | Glyma.13G101300.1 | Glyma.17G058400.1 | ||||||
| Glyma.15G167000.1 | MtSMXL13 | AT4G30350.1 | Zm00001eb412800_T002 | ESW26813 | Glyma.13G092700.1 | ||||
| Glyma.17G058400.1 | AT5G57710.1 | Zm00001eb257430_T002 | Glyma.17G067700.1 | ||||||
| MtSMXL4 | MtSMXL14 | AT2G29970.1 | ESW14200 | Glyma.02G226900.6 | |||||
| MtSMXL5 | AT1G07200.2 | ESW14200 | Glyma.02G226900.6 | ESW18766 | Glyma.11G174500.4 | ||||
| AT2G29970.1 | ESW18766 | Glyma.11G174500.4 | Glyma.14G193900.2 | ||||||
| Glyma.14G193900.2 | Glyma.18G062300.1 | ||||||||
| Glyma.18G062300.1 | MtSMXL15 | ||||||||
| MtSMXL6 | AT2G40130.2 | ESW13755 | Glyma.11G230700.1 | MtSMXL16 | AT5G15450.1 | ESW11501 | Glyma.08G242100.1 | ||
| ESW18766 | Glyma.18G026600.3 | ESW23286 | Glyma.13G058600.1 | ||||||
| ESW35546 | Glyma.18G062300.1 | Glyma.18G264400.1 | |||||||
| MtSMXL7 | AT5G51070.1 | Zm00001eb084420_T004 | ESW09825 | Glyma.04G203300.1 | Glyma.19G027900.1 | ||||
| Glyma.06G162200.1 | MtSMXL17 | AT3G48870.3 | Zm00001eb084890_T001 | ESW09858 | Glyma.04G200400.8 | ||||
| MtSMXL8 | AT5G50920.1 | ESW31965 | Glyma.05G201100.4 | ||||||
| MtSMXL9 | Glyma.06G165200.6 | ||||||||
| MtSMXL10 | AT2G25140.1 | Zm00001eb234160_T001 | ESW08954 | Glyma.04G062200.1 | Glyma.08G008400.1 |
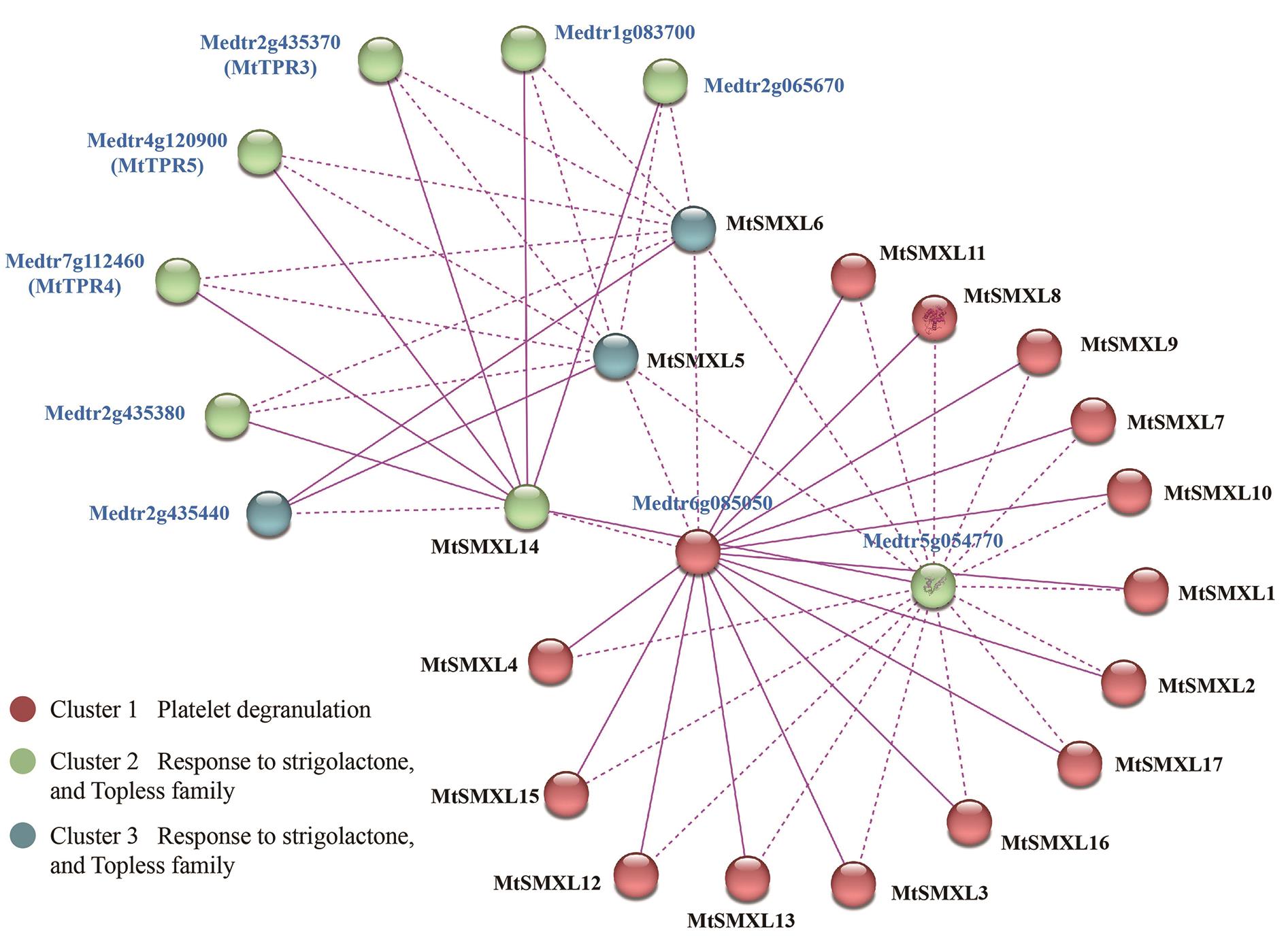
图7 MtSMXLs互作蛋白预测网络
Fig. 7 Putative network for MtSMXLs interacting proteins M. truncatula
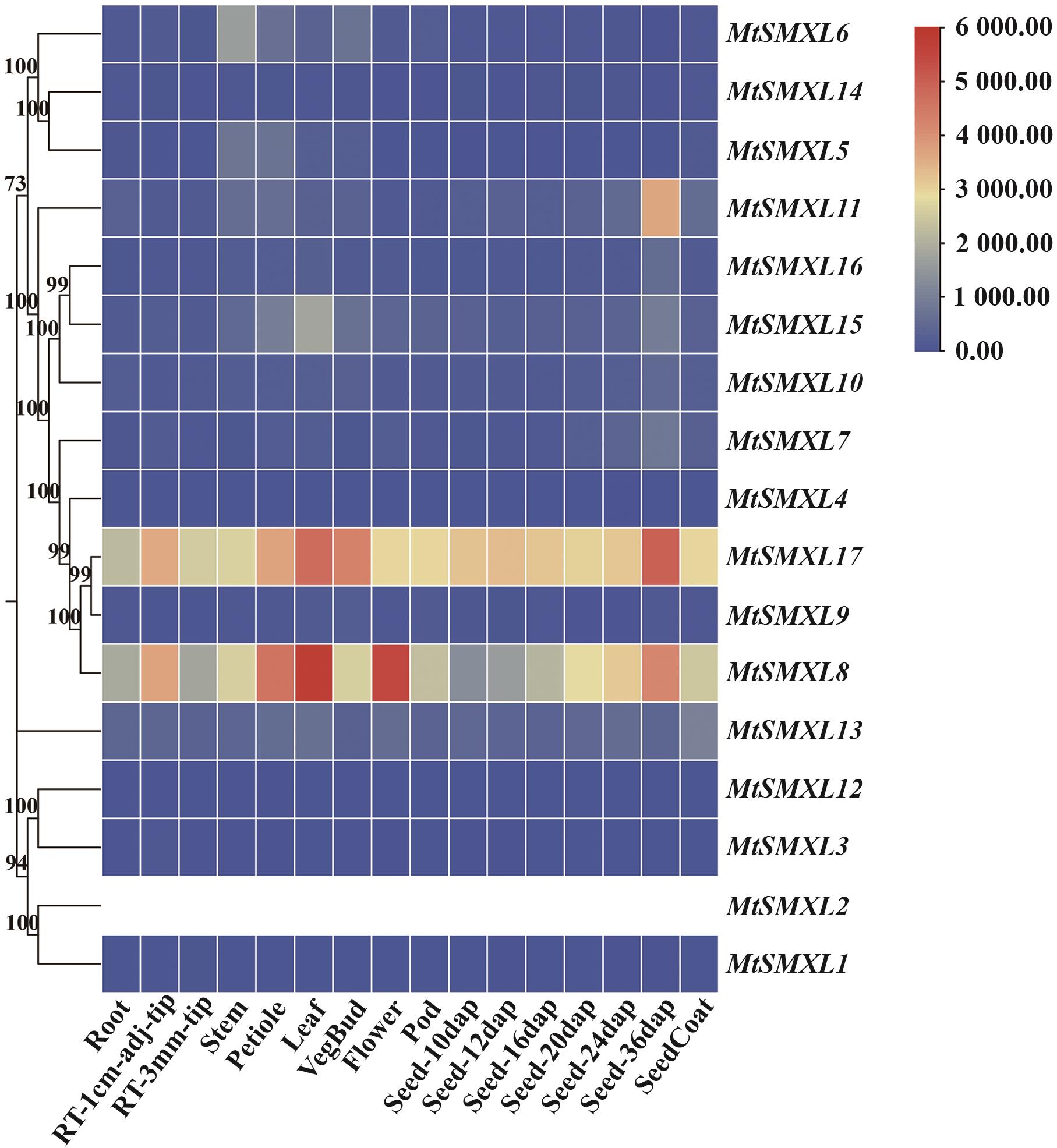
图8 蒺藜苜蓿SMXL家族基因不同组织部位表达分析右上角显示MtSMXL基因在不同组织中的表达量变化范围,红色越深代表表达量越高,蓝色越深代表表达量越低;图中adj指距离,dap指授粉后日数
Fig. 8 Expression analysis of SMXL family genes indifferent tissues of M. truncatulaThe upper right corner shows the expression range of the MtSMXL gene in different tissues. Darker red indicates higher expression, and darker blue indicates lower expression. In the figure, adj refers to distance, and dap refers to days after pollination
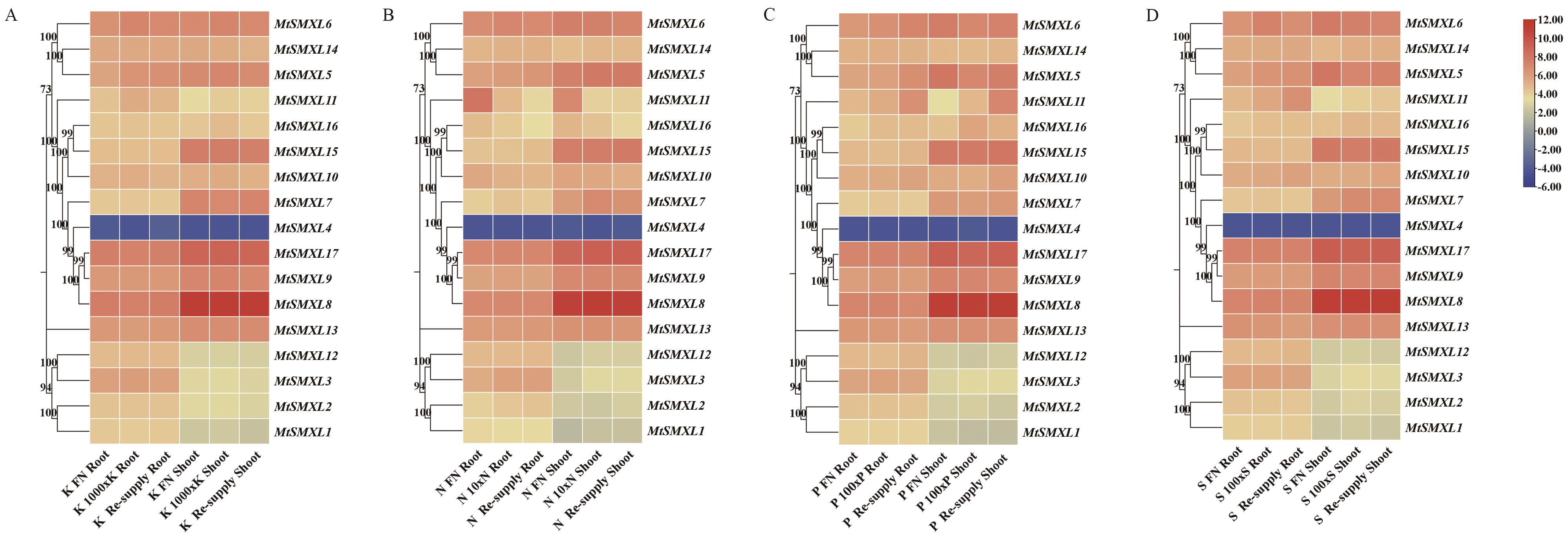
图9 蒺藜苜蓿MtSMXL成员在N、P、K和S元素缺乏条件下基因表达分析A:MtSMXLs基因响应低钾处理在根及芽中的表达水平热图;B:MtSMXLs基因响应低氮处理在根及芽中的表达水平热图;C:MtSMXLs基因响应低磷处理在根及芽中的表达水平热图;D:MtSMXLs基因响应低硫处理在根及芽中的表达水平热图
Fig. 9 Expression analysis of SMXL family genes of M. truncatula under conditions of N, P, K and S deficiencyA: Heatmap of the expressions of MtSMXLs genes in response to low potassium treatment in the roots and shoots. B: Heatmap of the expressions of MtSMXLs genes in response to low nitrogen treatment in the roots and shoots. C: Heatmap of the expressions of MtSMXLs genes in response to low phosphorus treatment in the roots and shoots. D: Heatmap of the expressions of MtSMXLs gene in response to low sulfur treatment in the roots and shoots
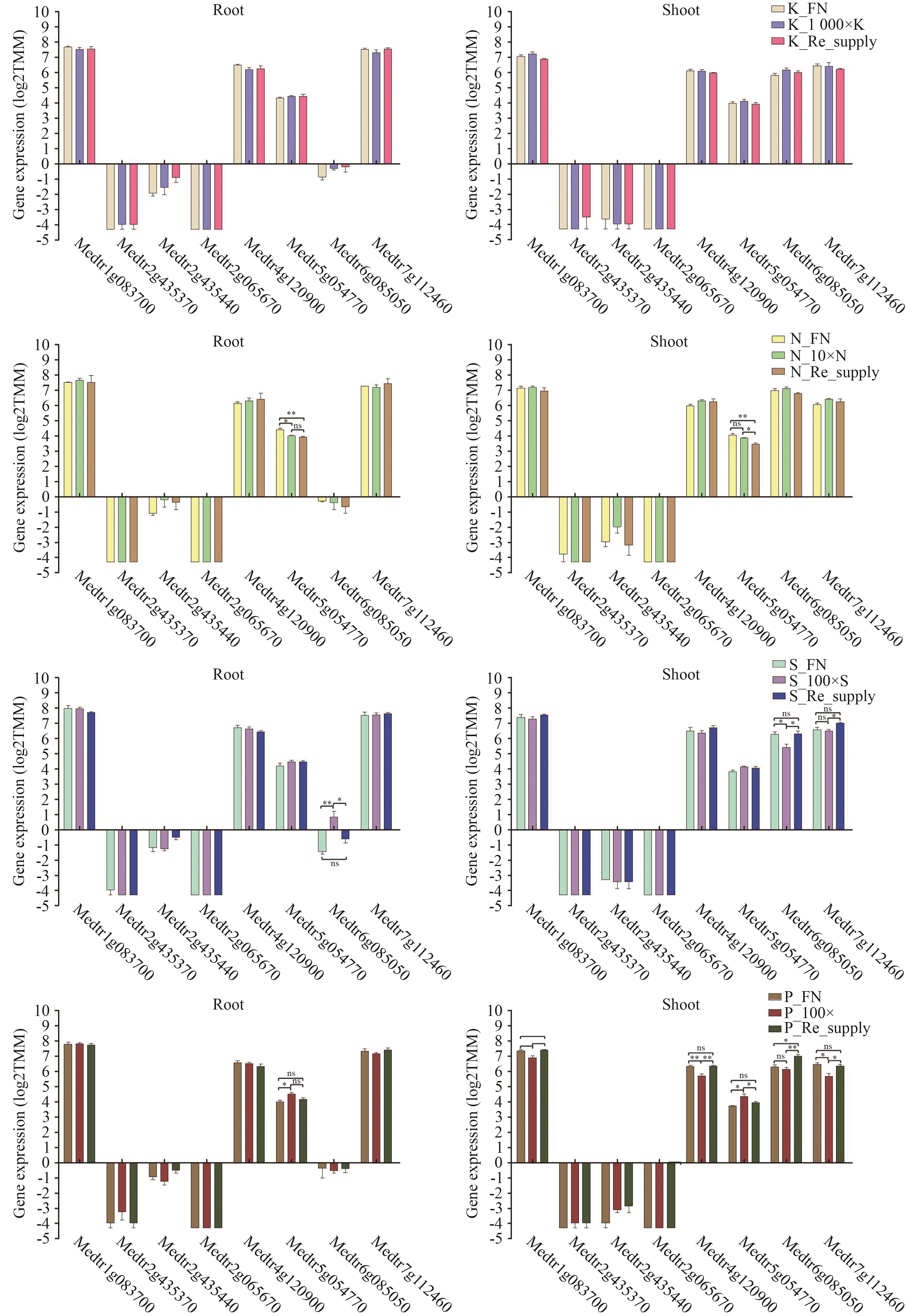
图10 候选互作蛋白编码基因在N、P、K和S处理下基因表达分析依据GraphPad Prism 5.0分析,*P<0.05,**P<0.005
Fig. 10 Gene expression analysis of candidate genes encoding interacting proteins under N, P, K and S treatmentsBased on GraphPad Prism 5.0 analysis, *P<0.05, **P<0.005
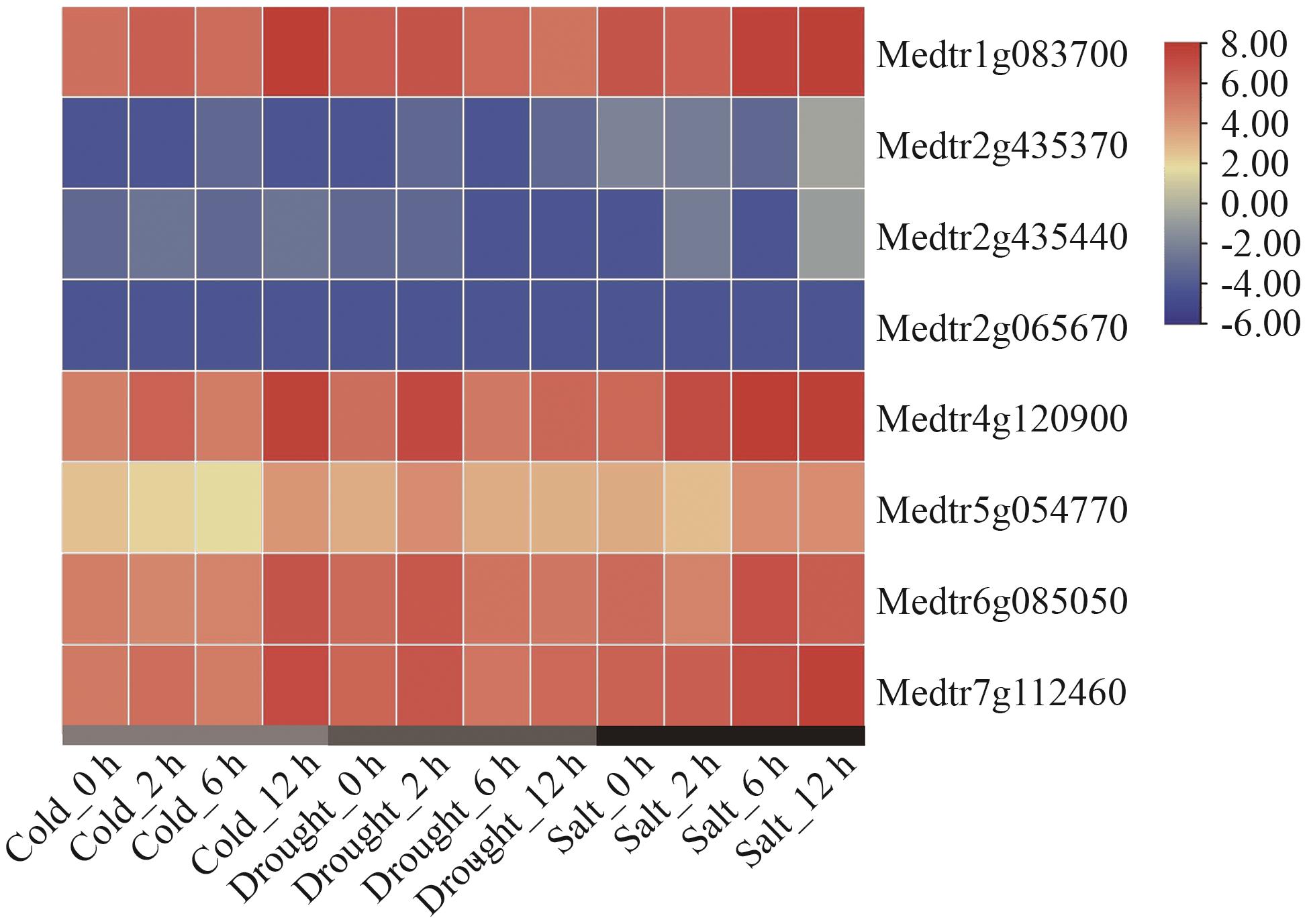
图11 候选互作蛋白编码基因在干旱、冷、盐处理条件下基因表达分析
Fig. 11 Gene expression analysis of candidate genes encoding interacting proteins under drought, cold and salt treatment conditions
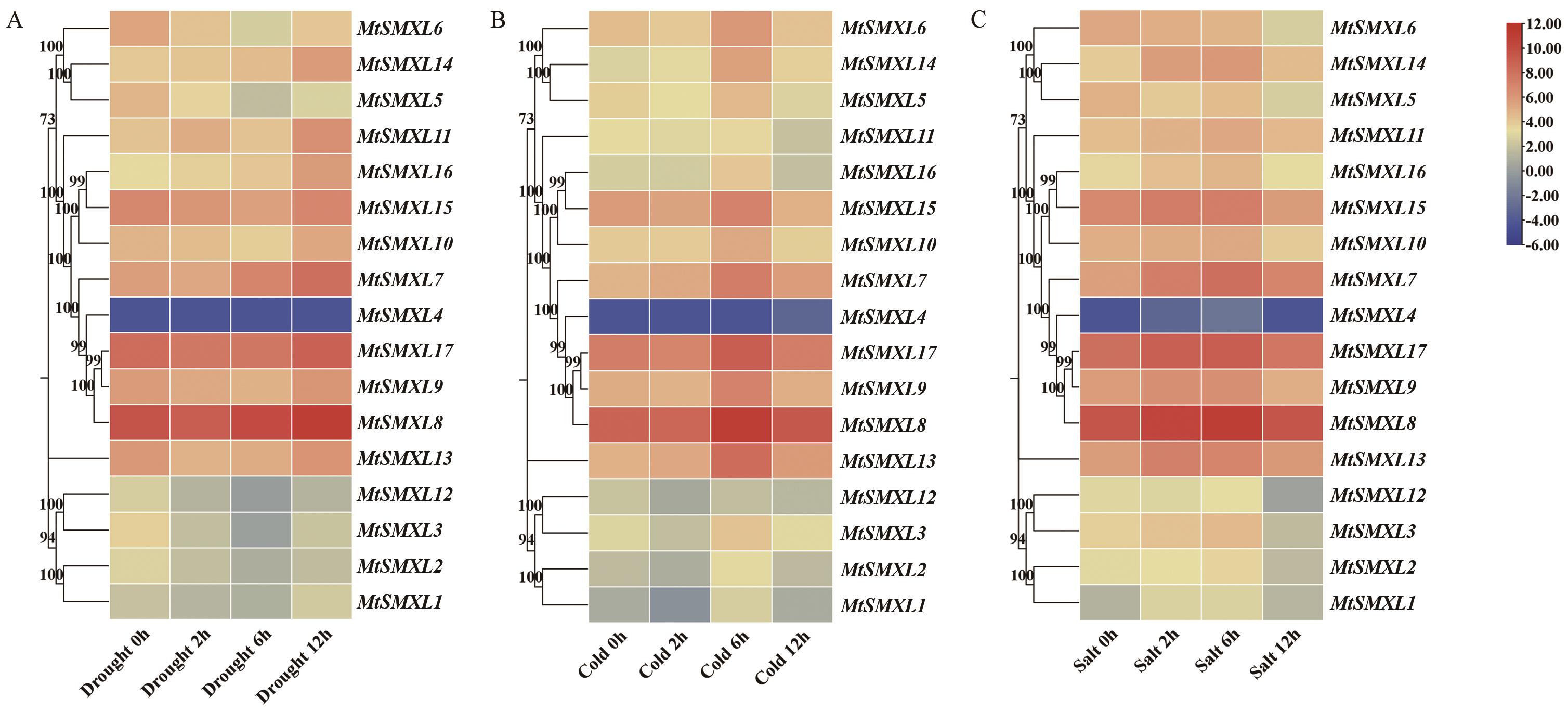
图12 蒺藜苜蓿MtSMXL成员在干旱、冷、盐处理条件下基因表达分析A:MtSMXLs基因在干旱处理下在幼苗中的表达水平热图;B:MtSMXLs基因在冷处理下在幼苗中的表达水平热图;C:MtSMXLs基因在盐处理下在幼苗中的表达水平热图
Fig. 12 Analysis of the gene expression patterns of SMXL family genes under drought, cold and salt treatmentA: Heatmap of the expression levels of MtSMXLs gene under drought treatment in seedlings. B: Heatmap of the expression levels of MtSMXLs gene under cold treatment in seedlings. C: Heatmap of the expression levels of MtSMXLs gene under salt treatment in seedlings
| [1] | Ma B, Zhu JB, Huang XZ. Diversification of plant SUPPRESSOR OF MAX2 1 (SMAX1)-like genes and genome-wide identification and characterization of cotton SMXL gene family [J]. BMC Plant Biol, 2023, 23(1): 419. |
| [2] | 卢昱帆, 闫怡秀, 秦颖杭, 等. 玉米SMXL基因家族的全基因组鉴定及在非生物胁迫中的潜在功能分析 [J/OL]. 分子植物育种, 2024: 1-19. |
| Lu YF, Yan YX, Qin YH, et al. Genome-wide identification and potential function analysis of maize SMXL gene family under abiotic stress [J/OL]. Mol Plant Breed, 2024: 1-19. | |
| [3] | Bhoi A, Yadu B, Chandra J, et al. Contribution of strigolactone in plant physiology, hormonal interaction and abiotic stresses [J]. Planta, 2021, 254(2): 28. |
| [4] | Wang L, Wang B, Yu H, et al. Transcriptional regulation of strigolactone signalling in Arabidopsis [J]. Nature, 2020, 583(7815): 277-281. |
| [5] | 王雪菱, 王如月, 李际红, 等. 独脚金内酯抑制因子D53/SMXLs基因的研究进展 [J]. 农学学报, 2023, 13(10): 37-43. |
| Wang XL, Wang RY, Li JH, et al. The strigolactones inhibitory factor D53/SMXLs: a review [J]. J Agric, 2023, 13(10): 37-43. | |
| [6] | Arite T, Umehara M, Ishikawa S, et al. d14, a strigolactone-insensitive mutant of rice, shows an accelerated outgrowth of tillers [J]. Plant Cell Physiol, 2009, 50(8): 1416-1424. |
| [7] | Jiang L, Liu X, Xiong GS, et al. DWARF 53 acts as a repressor of strigolactone signalling in rice [J]. Nature, 2013, 504(7480): 401-405. |
| [8] | Stanga JP, Smith SM, Briggs WR, et al. SUPPRESSOR OF MORE AXILLARY GROWTH2 1 controls seed germination and seedling development in Arabidopsis [J]. Plant Physiol, 2013, 163(1): 318-330. |
| [9] | Yao RF, Ming ZH, Yan LM, et al. DWARF14 is a non-canonical hormone receptor for strigolactone [J]. Nature, 2016, 536(7617): 469-473. |
| [10] | Wang L, Wang B, Jiang L, et al. Strigolactone signaling in Arabidopsis regulates shoot development by targeting D53-like SMXL repressor proteins for ubiquitination and degradation [J]. Plant Cell, 2015, 27(11): 3128-3142. |
| [11] | Wallner ES, López-Salmerón V, Belevich I, et al. Strigolactone- and karrikin-independent SMXL proteins are central regulators of phloem formation [J]. Curr Biol, 2017, 27(8): 1241-1247. |
| [12] | Zhou F, Lin QB, Zhu LH, et al. Correction: Corrigendum: D14-SCFD3-dependent degradation of D53 regulates strigolactone signalling [J]. Nature, 2016, 532(7599): 402. |
| [13] | Wu YY, Hou BH, Lee WC, et al. DCL2- and RDR6-dependent transitive silencing of SMXL4 and SMXL5 in Arabidopsis dcl4 mutants causes defective phloem transport and carbohydrate over-accumulation [J]. Plant J, 2017, 90(6): 1064-1078. |
| [14] | Stanga JP, Morffy N, Nelson DC. Functional redundancy in the control of seedling growth by the karrikin signaling pathway [J]. Planta, 2016, 243(6): 1397-1406. |
| [15] | Villaécija-Aguilar JA, Hamon-Josse M, Carbonnel S, et al. SMAX1/SMXL2 regulate root and root hair development downstream of KAI2-mediated signalling in Arabidopsis [J]. PLoS Genet, 2019, 15(8): e1008327. |
| [16] | Wang ZJ, Jiang ZN, Wan HP, et al. Genome-wide identification of the SMXL gene family in common wheat and expression analysis of TaSMXLs under abiotic stress [J]. Agronomy, 2025, 15(3): 656. |
| [17] | 贾婷婷, 竺丽萍, 肖光辉, 等. 棉花SMXL基因家族的全基因组分析及表达分析 [J]. 中国科学: 生命科学, 2022, 52(12): 1868-1882. |
| Jia TT, Zhu LP, Xiao GH, et al. Genome-wide identification and expression analysis of the SMXL gene family in cotton [J]. Sci Sin Vitae, 2022, 52(12): 1868-1882. | |
| [18] | Zhang H, Wang L, Gao Y, et al. Genome-wide identification of SMXL gene family in soybean and expression analysis of GmSMXLs under shade stress [J]. Plants, 2022, 11(18): 2410. |
| [19] | Fu XJ, Wang J, Shangguan TW, et al. SMXLs regulate seed germination under salinity and drought stress in soybean [J]. Plant Growth Regul, 2022, 96(3): 397-408. |
| [20] | 陈莹, 张春渝, 许小琼, 等. 4种无患子科植物的SMXL家族全基因组鉴定、进化及DlSMXLs在龙眼体胚发生过程中的表达分析 [J]. 东南园艺, 2024, 12(1): 17-29. |
| Chen Y, Zhang CY, Xu XQ, et al. Genome-wide identification and evolutionary analysis of the SMXL gene family in four Sapindaceae species and expression analysis of DlSMXLs during somatic embryogenesis in Longan [J]. Southeast Hortic, 2024, 12(1): 17-29. | |
| [21] | Li R, An JP, You CX, et al. Genome-wide analysis and identification of the SMXL gene family in apple (Malus×domestica) [J]. Tree Genet Genomes, 2018, 14(4): 61. |
| [22] | 孙卫健. 苹果独脚金内酯信号途径阻遏蛋白同源基因MdSMXL8.2的功能鉴定 [D]. 泰安: 山东农业大学, 2020. |
| Sun WJ. Functional identification of apple strigolactone signaling pathway repressor homologous gene MdSMXL8.2 [D]. Tai’an: Shandong Agricultural University, 2020. | |
| [23] | Basso MF, Contaldi F, Lo Celso F, et al. Identification and expression profile of the SMAX/SMXL family genes in chickpea and lentil provide important players of biotechnological interest involved in plant branching [J]. Planta, 2023, 259(1): 1. |
| [24] | Yuan S, Zhang WL, Zhang YX. Characterization of SUPPRESSOR OF MAX2 1-LIKE (SMXL) genes in ‘Duli’ (Pyrus betulifolia L.) and expression analysis of PbSMXLs in response to plant growth regulators and salt stress [J]. Agronomy, 2024, 14(12): 2778. |
| [25] | Fang PP, Li MX, Guo QW, et al. Genome-wide analysis of the SMXL gene family in common bean and identification of karrikin-responsive PvSMXL2 as a negative regulator of PEG-induced drought stress [J]. Gene, 2023, 887: 147741. |
| [26] | Sun MT, Wang DY, Liu CS, et al. Genome-wide identification and analysis of the SUPPRESSOR of MAX2 1-LIKE gene family and its interaction with DWARF14 in poplar [J]. BMC Plant Biol, 2023, 23(1): 105. |
| [27] | Moturu TR, Thula S, Singh RK, et al. Molecular evolution and diversification of the SMXL gene family [J]. J Exp Bot, 2018, 69(9): 2367-2378. |
| [28] | Yang TJ, Kim JS, Kwon SJ, et al. Sequence-level analysis of the diploidization process in the triplicated FLOWERING LOCUS C region of Brassica rapa [J]. Plant Cell, 2006, 18(6): 1339-1347. |
| [29] | de Bang TC, Lundquist PK, Dai XB, et al. Genome-wide identification of Medicago Peptides involved in macronutrient responses and nodulation [J]. Plant Physiol, 2017, 175(4): 1669-1689. |
| [30] | Hu B, Wu H, Huang WF, et al. SWEET gene family in Medicago truncatula: genome-wide identification, expression and substrate specificity analysis [J]. Plants, 2019, 8(9): 338. |
| [31] | Qiao X, Li QH, Yin H, et al. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants [J]. Genome Biol, 2019, 20(1): 38. |
| [32] | 贺红利, 朴京培, 孙嘉囡, 等. 蒺藜苜蓿CIPK基因家族全基因组鉴定及表达分析 [J]. 中国草地学报, 2021, 43(9): 1-13. |
| He HL, Piao JP, Sun JN, et al. Genome-wide identification and expression analysis of CIPK gene family in Medicago truncatula [J]. Chin J Grassland, 2021, 43(9): 1-13. | |
| [33] | Wolfe KH, Gouy M, Yang YW, et al. Date of the monocot-dicot divergence estimated from chloroplast DNA sequence data [J]. Proc Natl Acad Sci U S A, 1989, 86(16): 6201-6205. |
| [34] | Zhao YY, Zhang R, Jiang KW, et al. Nuclear phylotranscriptomics and phylogenomics support numerous polyploidization events and hypotheses for the evolution of rhizobial nitrogen-fixing symbiosis in Fabaceae [J]. Mol Plant, 2021, 14(5): 748-773. |
| [35] | Zhang L, Wu RL, Mur LAJ, et al. Assembly of high-quality genomes of the locoweed Oxytropis ochrocephala and its endophyte Alternaria oxytropis provides new evidence for their symbiotic relationship and swainsonine biosynthesis [J]. Mol Ecol Resour, 2023, 23(1): 253-272. |
| [36] | 张孝廉, 张吉顺, 雷波, 等. 植物MLO蛋白研究进展 [J]. 植物生理学报, 2018, 54(7): 1159-1171. |
| Zhang XL, Zhang JS, Lei B, et al. Research progress of plant MLO protein [J]. Plant Physiol Commun, 2018, 54(7): 1159-1171. | |
| [37] | Kim MY, Kang YJ, Lee T, et al. Divergence of flowering-related genes in three legume species [J]. Plant Genome, 2013, 6(3): plantgenome2013.03.0008. |
| [38] | Liang YY, Ward S, Li P, et al. SMAX1-LIKE7 signals from the nucleus to regulate shoot development in Arabidopsis via partially EAR motif-independent mechanisms [J]. Plant Cell, 2016, 28(7): 1581-1601. |
| [39] | Fang JJ, Guo TT, Xie ZW, et al. The URL1-ROC5-TPL2 transcriptional repressor complex represses the ACL1 gene to modulate leaf rolling in rice [J]. Plant Physiol, 2021, 185(4): 1722-1744. |
| [1] | 高飞, 张宇曦, 穆頔, 陈峥, 陈洪艳. 鼠源罗伊氏黏液乳杆菌的分离鉴定及全基因组测序分析[J]. 生物技术通报, 2026, 42(7): 1-13. |
| [2] | 张驰昊, 刘晋囡, 晁跃辉. 蒺藜苜蓿bZIP转录因子MtbZIP29的克隆及功能分析[J]. 生物技术通报, 2026, 42(1): 241-250. |
| [3] | 刘长命, 张新悦, 杨昕萌, 刘阳, 李玥涵, 汪可清. 一株新的魔芋软腐病致病菌的生物学特性及基因组分析[J]. 生物技术通报, 2026, 42(1): 294-304. |
| [4] | 吕镇, 甘恬, 霍思羽, 赵晨笛, 赵梦瑶, 李亚涛, 马玉超, 耿玉清. 产Surfactin贝莱斯芽胞杆菌C5A-1的鉴定和所产Surfactin对植物的促生效果[J]. 生物技术通报, 2025, 41(9): 265-276. |
| [5] | 程婷婷, 刘俊, 王利丽, 练从龙, 魏文君, 郭辉, 吴尧琳, 杨晶凡, 兰金旭, 陈随清. 杜仲查尔酮异构酶基因家族全基因组鉴定及其表达模式分析[J]. 生物技术通报, 2025, 41(9): 242-255. |
| [6] | 李亚涛, 张志鹏, 赵梦瑶, 吕镇, 甘恬, 魏浩, 吴书凤, 马玉超. 根瘤菌Bd1的全基因组分析及TetR3对细胞生长和结瘤的负调控功能[J]. 生物技术通报, 2025, 41(9): 289-301. |
| [7] | 魏瑶, 张晶晶, 崔云晓, 刘钰, 刘海瑞. 忍冬属忍冬组植物叶绿体基因组进化分析[J]. 生物技术通报, 2025, 41(8): 276-288. |
| [8] | 李凯月, 邓晓霞, 殷缘, 杜亚彤, 徐元静, 王竞红, 于耸, 蔺吉祥. 蓖麻LEA基因家族的鉴定和铝胁迫响应分析[J]. 生物技术通报, 2025, 41(7): 128-138. |
| [9] | 龚钰涵, 陈兰, 尚方慧子, 郝灵颖, 刘硕谦. 茶树TRB基因家族鉴定及表达模式分析[J]. 生物技术通报, 2025, 41(7): 214-225. |
| [10] | 张津浩, 邓辉, 张清壮, 陶禹, 周池, 李鑫. 贝莱斯芽胞杆菌XY40-1对百合球茎生长、品质及镉含量的调控作用[J]. 生物技术通报, 2025, 41(7): 281-291. |
| [11] | 周熠, 刘勇波. 基因组进化过程中的基因丢失机制与功能研究进展[J]. 生物技术通报, 2025, 41(6): 38-48. |
| [12] | 李晨莹, 孔大帅, 李若楠, 张玉波, 阎萍, 李奎, 孔思远. 前沿组学技术创新助力畜禽生物育种[J]. 生物技术通报, 2025, 41(6): 71-86. |
| [13] | 霍贯中, 张欣濡, 田士军, 李君. CRISPR/Cas12a基因编辑技术在植物中的研究进展[J]. 生物技术通报, 2025, 41(6): 1-11. |
| [14] | 安昌, 徐文波, 陆琳, 李登麟, 姚艺新, 林彦翔, 杨成梓, 秦源, 郑平. 不同产地珊瑚菜叶绿体基因组特征的差异性研究[J]. 生物技术通报, 2025, 41(6): 229-242. |
| [15] | 吴泽银, 黄晨阳, 赵梦然, 张利姣, 姚方杰. 短柄白黄侧耳CCMSSC 04611基因组特异性分析[J]. 生物技术通报, 2025, 41(5): 320-332. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||