







生物技术通报 ›› 2022, Vol. 38 ›› Issue (9): 180-190.doi: 10.13560/j.cnki.biotech.bull.1985.2022-0344
司诚1,2( ), 钟启文1, 杨世鹏1()
), 钟启文1, 杨世鹏1()
收稿日期:2022-03-22
出版日期:2022-09-26
发布日期:2022-10-11
作者简介:司诚,女,硕士研究生,研究方向:作物遗传与生长发育;E-mail: 基金资助:
SI Cheng1,2(), ZHONG Qi-wen1, YANG Shi-peng1()
Received:2022-03-22
Published:2022-09-26
Online:2022-10-11
摘要:
香瓜茄又名人参果,具有抗氧化、抗肿瘤、抗糖尿病等多种生物活性。为丰富茄科作物基因组信息及进化发育历程,获取香瓜茄全基因组序列信息,同时为香瓜茄相关分子研究奠定基础。以香瓜茄植物组织为试验材料,基于Illumina HiSeq构建小片段文库进行基因组特征评估,利用PacBio三代测序技术、Hi-C技术构建及组装香瓜茄全基因组数据库。利用生物信息学方法对获得的基因组序列进行组装、功能注释以及进化分析研究。结果表明,获得54.11 Gb Illumina HiSeq数据;获得55.08 Gb PacBio数据,reads平均长度为14 179 bp;获得Hi-C数据量约143 Gb;拼接得到该基因组contig序列总长为1.16 Gb,Hi-C纠错后contig N50为22.63 Mb;Hi-C挂载染色体,共有1.12 Gb长度的序列可以挂载到12条染色体上,占比97.16%;其中,能够确定顺序和方向的序列长度为1.08 Gb,占定位染色体序列总长度的96.11%,得到基因组大小1.25 Gb;预测有64.22%的重复序列,41 571个基因,99.06%的基因可以注释到NR、GO、KEGG等数据库中;预测得到4 360个tRNA、5 677个rRNA、154个miRNA;得到449个假基因。香瓜茄与马铃薯的进化时间大约在12.82 MYA。
司诚, 钟启文, 杨世鹏. 基于PacBio三代测序的香瓜茄(人参果)基因组的组装[J]. 生物技术通报, 2022, 38(9): 180-190.
SI Cheng, ZHONG Qi-wen, YANG Shi-peng. Assembly of Pepino Genome Based on PacBio's Third-generation Sequencing Technology[J]. Biotechnology Bulletin, 2022, 38(9): 180-190.
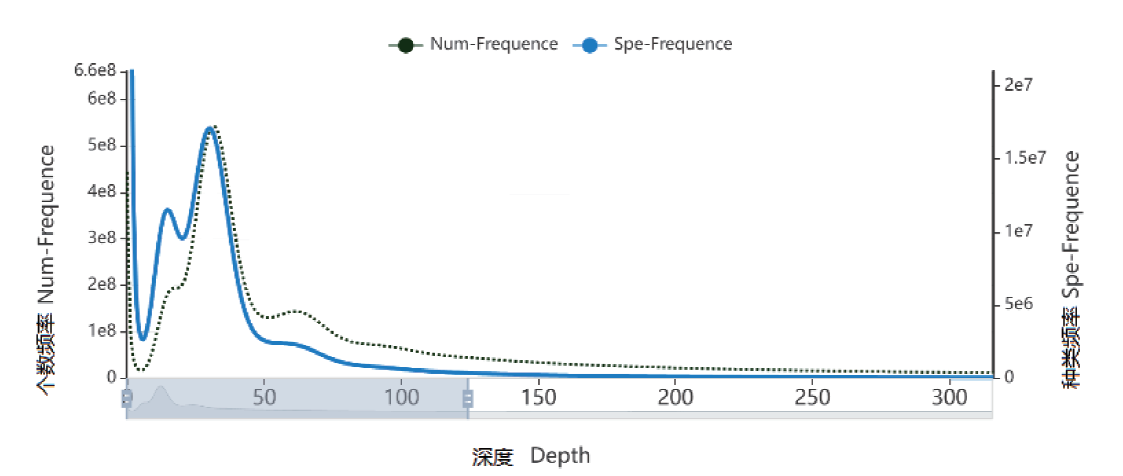
图1 Depth和K-mer个数及种类频率分布图
Fig. 1 Depth and number of K-mer as well species freque-ncy distribution
| K-mer | K-mer总数量Total K-mer number | K-mer深度K-mer depth | 基因组大小Genome size/Mb | 修正后基因组大小Corrected genome size/Mb | 杂合率Heterozygosity/% | 重复序列比例 Repeat sequence ratio/% |
|---|---|---|---|---|---|---|
| 6 | 38 824 569 291 | 31 | 1 252.41 | 1 238.06 | 0.84 | 65.87 |
表1 香瓜茄基因组特征
Table 1 Pepino genomic characteristics
| K-mer | K-mer总数量Total K-mer number | K-mer深度K-mer depth | 基因组大小Genome size/Mb | 修正后基因组大小Corrected genome size/Mb | 杂合率Heterozygosity/% | 重复序列比例 Repeat sequence ratio/% |
|---|---|---|---|---|---|---|
| 6 | 38 824 569 291 | 31 | 1 252.41 | 1 238.06 | 0.84 | 65.87 |
| 项目Item | 总长度Total length/bp | 总数量Total number | 最大长度Max length/bp | N50长度N50-length/bp | N90长度N90-length/bp |
|---|---|---|---|---|---|
| Contig | 1 141 353 553 | 1 952 498 | 125 830 | 2 049 | 165 |
| Scaffold | 1 169 596 440 | 1 679 956 | 191 961 | 3 185 | 199 |
表2 组装结果统计
Table 2 Statistics of assembly results
| 项目Item | 总长度Total length/bp | 总数量Total number | 最大长度Max length/bp | N50长度N50-length/bp | N90长度N90-length/bp |
|---|---|---|---|---|---|
| Contig | 1 141 353 553 | 1 952 498 | 125 830 | 2 049 | 165 |
| Scaffold | 1 169 596 440 | 1 679 956 | 191 961 | 3 185 | 199 |
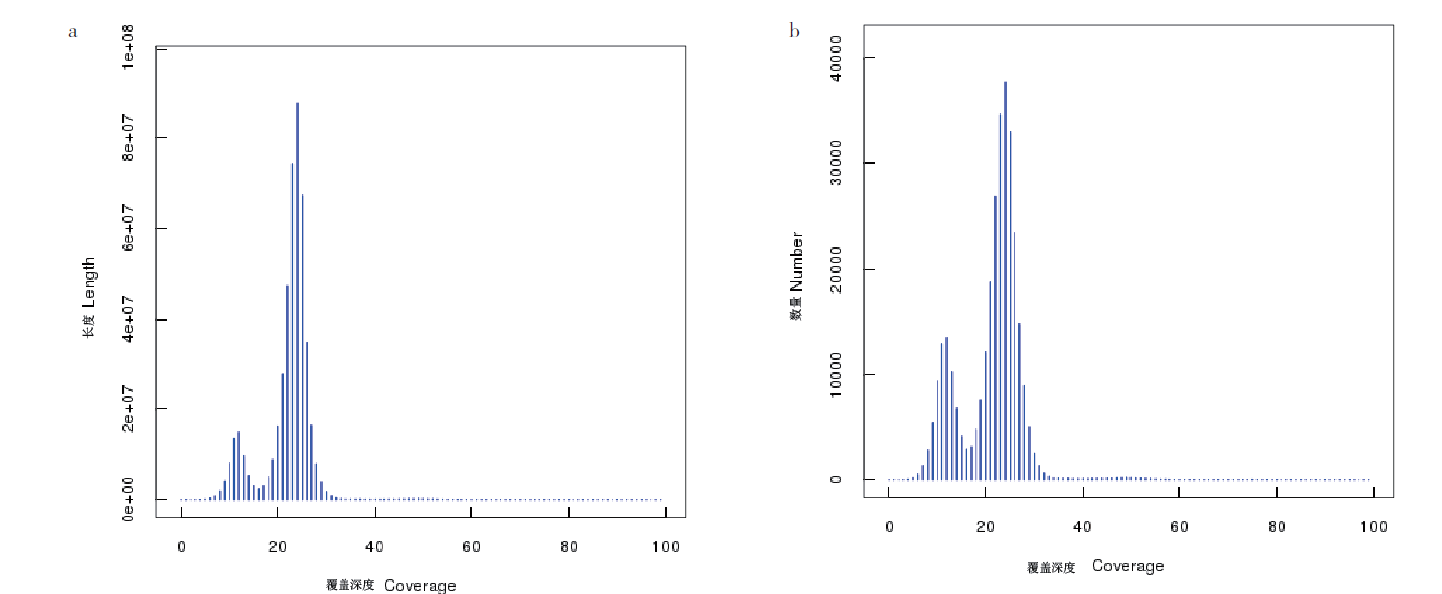
图2 Contig覆盖深度、长度和数量分布图 a:contig覆盖深度和长度分布图;b:contig覆盖深度和数量分布图
Fig. 2 Contig coverage depth, length and number distribution map a:Length distribution chart. b:Quantity distribution chart
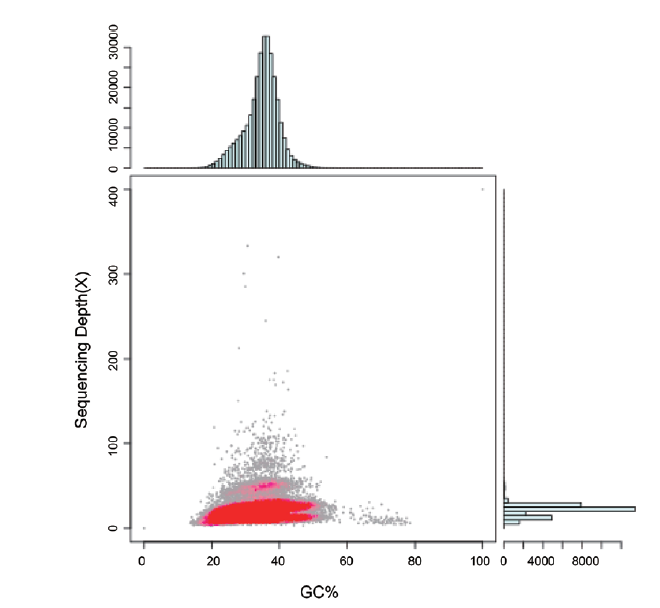
图3 GC含量与测序深度(depth)关联分析统计图
Fig. 3 Statistical analysis of GC content and sequencing depth
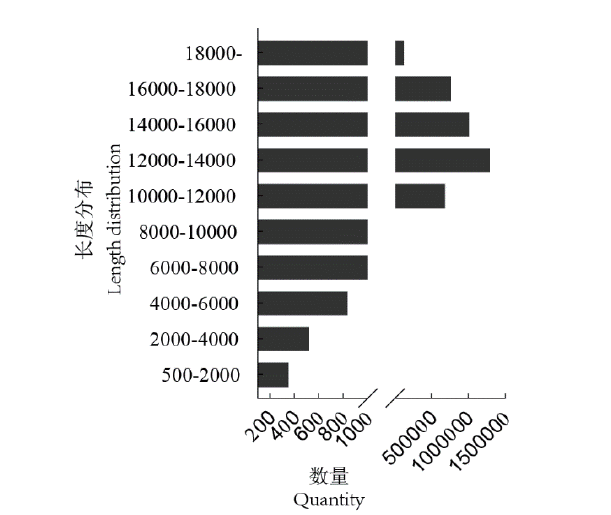
图4 Reads长度分布统计
Fig. 4 Reads length distribution statistics
| 重叠群数目 CtgNum | 重叠群长度 CtgLen/bp | 重叠群N50长度 CtgN50/bp | 重叠群N90长度 CtgN90/bp | 最长重叠群长度 CtgMax/bp | 碱基含量 GC/% |
|---|---|---|---|---|---|
| 1 813 | 1 156 071 317 | 22 628 432 | 596 645 | 83 851 337 | 35.83 |
表3 组装结果的统计信息
Table 3 Statistical information of assembly results
| 重叠群数目 CtgNum | 重叠群长度 CtgLen/bp | 重叠群N50长度 CtgN50/bp | 重叠群N90长度 CtgN90/bp | 最长重叠群长度 CtgMax/bp | 碱基含量 GC/% |
|---|---|---|---|---|---|
| 1 813 | 1 156 071 317 | 22 628 432 | 596 645 | 83 851 337 | 35.83 |
| 项目Item | 基因组信息Genome information |
|---|---|
| 总数TotalNum | 1 566 |
| 最小长度MinLen | 1 000 |
| Scaffold的数量ScfNum | 1 566 |
| Scaffold的长度ScfLen/bp | 1 156 096 017 |
| Scaffold N50长度ScfN50/bp | 87 253 278 |
| Scaffold N90长度ScfN90/bp | 74 267 810 |
| 最长的scaffold ScfMax/bp | 111 830 586 |
| Scaffold数目mScfNum | 0 |
| Scaffold的长度mScfLen | 0 |
| Contig数目CtgNum | 1 813 |
| Ctg长度CtgLen/bp | 1 156 071 317 |
| Contig N50的长度CtgN50/bp | 22 628 432 |
| Contig N90的长度CtgN90/bp | 596 645 |
| 最长的contig CtgMax/bp | 83 851 337 |
| GC含量GC/% | 35.83 |
| Contig数目GapNum | 247 |
| Gap长度GapLen/bp | 24 700 |
| 最大的gap MaxGap | 100 |
表4 香瓜茄Hi-C组装的基因组信息
Table 4 Hi-C assembly information of the pepino genome
| 项目Item | 基因组信息Genome information |
|---|---|
| 总数TotalNum | 1 566 |
| 最小长度MinLen | 1 000 |
| Scaffold的数量ScfNum | 1 566 |
| Scaffold的长度ScfLen/bp | 1 156 096 017 |
| Scaffold N50长度ScfN50/bp | 87 253 278 |
| Scaffold N90长度ScfN90/bp | 74 267 810 |
| 最长的scaffold ScfMax/bp | 111 830 586 |
| Scaffold数目mScfNum | 0 |
| Scaffold的长度mScfLen | 0 |
| Contig数目CtgNum | 1 813 |
| Ctg长度CtgLen/bp | 1 156 071 317 |
| Contig N50的长度CtgN50/bp | 22 628 432 |
| Contig N90的长度CtgN90/bp | 596 645 |
| 最长的contig CtgMax/bp | 83 851 337 |
| GC含量GC/% | 35.83 |
| Contig数目GapNum | 247 |
| Gap长度GapLen/bp | 24 700 |
| 最大的gap MaxGap | 100 |

图5 香瓜茄基因组Hi-C组装染色体交互热图 横坐标、纵坐标均代表每个bin在相应染色体群组上的Order
Fig. 5 Hi-C assembly chromosome interaction heat map of pepino genome Both horizontal and vertical coordinates represent the Order of each bin on the corresponding chromosome group
| 方法 Method | 软件 Software | 物种 Species | 基因数目 Gene number |
|---|---|---|---|
| Ab initio | Augustus | 26 025 | |
| SNAP | 47 891 | ||
| Homology-based | GeMoMa | 拟南芥A. thaliana | 26 652 |
| 辣椒C. annuum | 37 150 | ||
| 番茄S. lycopersicum | 32 312 | ||
| 潘那利番茄 S. pennellii | 33 755 | ||
| 马铃薯S. tuberosum | 51 586 | ||
| RNAseq | GeneMarkS-T | 19 020 | |
| PASA | 12 454 | ||
| Integration | EVM | 41 571 |
表5 香瓜茄基因预测结果
Table 5 Prediction results of pepino gene
| 方法 Method | 软件 Software | 物种 Species | 基因数目 Gene number |
|---|---|---|---|
| Ab initio | Augustus | 26 025 | |
| SNAP | 47 891 | ||
| Homology-based | GeMoMa | 拟南芥A. thaliana | 26 652 |
| 辣椒C. annuum | 37 150 | ||
| 番茄S. lycopersicum | 32 312 | ||
| 潘那利番茄 S. pennellii | 33 755 | ||
| 马铃薯S. tuberosum | 51 586 | ||
| RNAseq | GeneMarkS-T | 19 020 | |
| PASA | 12 454 | ||
| Integration | EVM | 41 571 |
| 功能注释数据库Function-annotated database | 注释到的基因数目Number of annotated genes | 占总数据库的百分比Percentage in database /% |
|---|---|---|
| GO_Annotation | 30 713 | 73.88 |
| KEGG_Annotation | 29 114 | 70.03 |
| KOG_Annotation | 20 636 | 49.64 |
| Pfam_Annotation | 31 802 | 76.5 |
| Swissprot_Annotation | 28 876 | 69.46 |
| TrEMBL_Annotation | 41 085 | 98.83 |
| eggNOG_Annotation | 32 433 | 78.02 |
| nr_Annotation | 40 785 | 98.11 |
| All_Annotated | 41 179 | 99.06 |
表6 香瓜茄基因功能注释统计信息
Table 6 Statistical information of pepino gene function annotation
| 功能注释数据库Function-annotated database | 注释到的基因数目Number of annotated genes | 占总数据库的百分比Percentage in database /% |
|---|---|---|
| GO_Annotation | 30 713 | 73.88 |
| KEGG_Annotation | 29 114 | 70.03 |
| KOG_Annotation | 20 636 | 49.64 |
| Pfam_Annotation | 31 802 | 76.5 |
| Swissprot_Annotation | 28 876 | 69.46 |
| TrEMBL_Annotation | 41 085 | 98.83 |
| eggNOG_Annotation | 32 433 | 78.02 |
| nr_Annotation | 40 785 | 98.11 |
| All_Annotated | 41 179 | 99.06 |
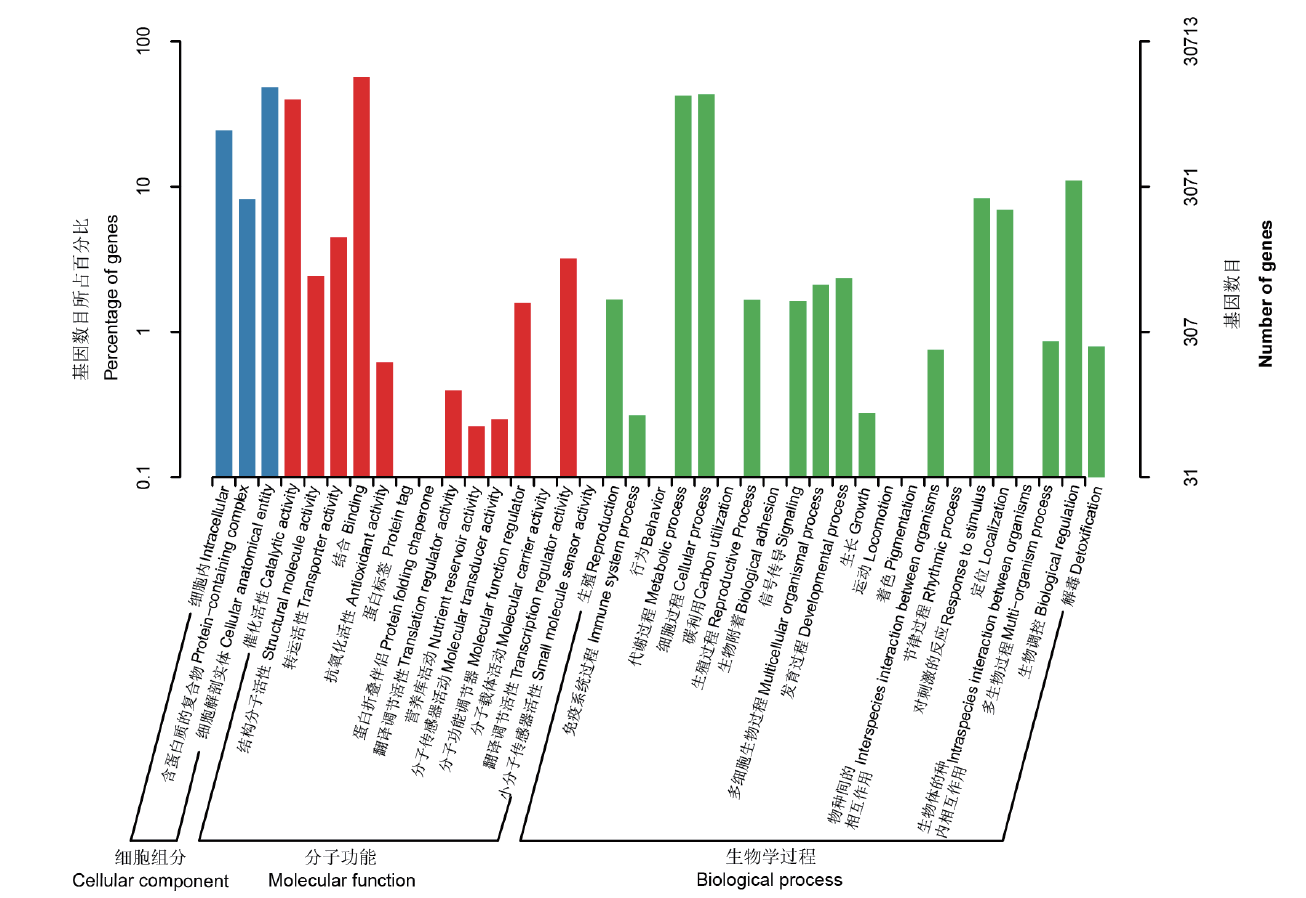
图6 GO二级节点注释分类统计图
Fig. 6 Statistical chart of GO secondary node annotation classification
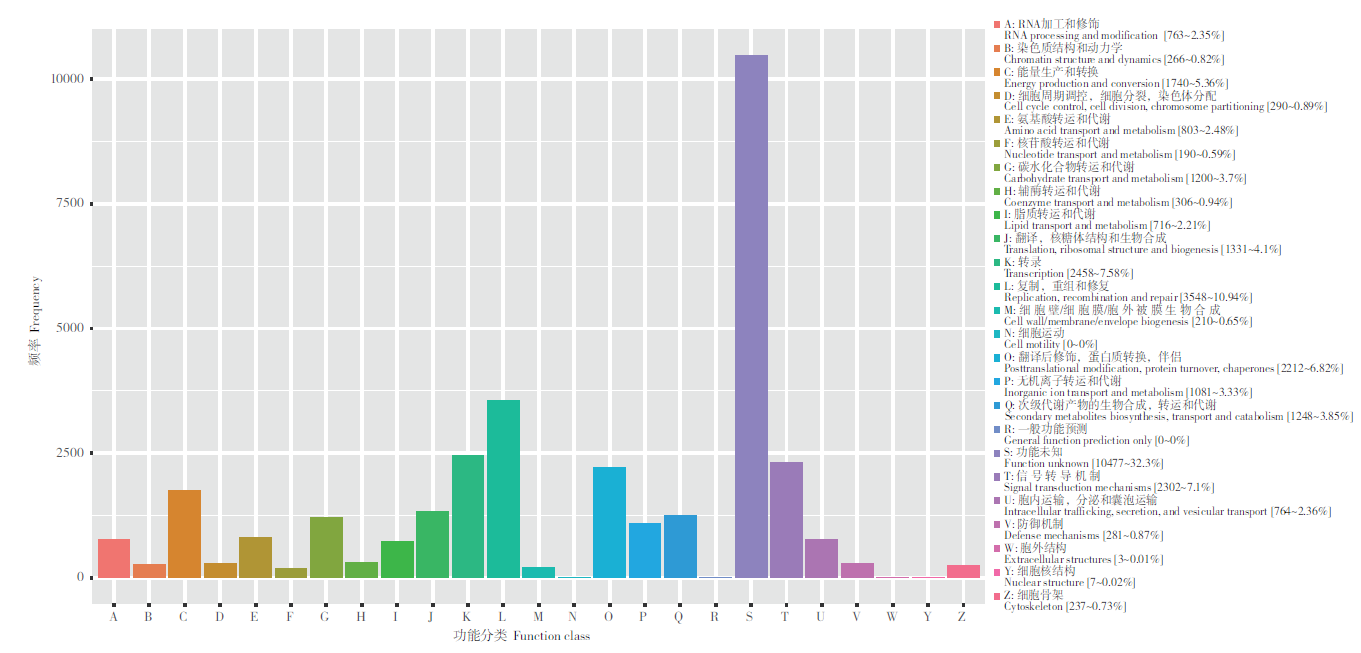
图7 eggNog功能注释分类统计图
Fig. 7 eggNog functional annotation classification
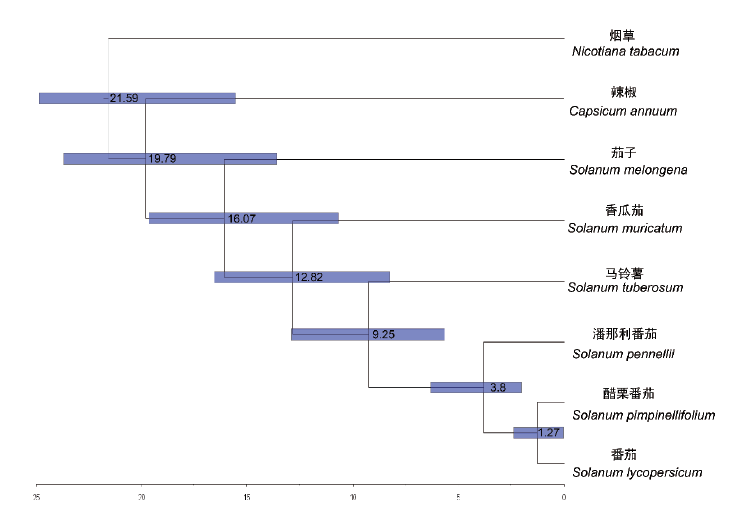
图8 物种间分化时间
Fig. 8 Differentiation time between species
| [1] | Pandey S, Prakash S, Prasad Y, et al. Studies on the effects of different surface sterilization agents under in vitro culture of pepino(Solanum muricatum Ait. )cv[J]. Valentia, 2021, 10(6):371-377. |
| [2] |
Fribourg CE, Gibbs AJ, Adams IP, et al. Biological and molecular properties of wild potato mosaic virus isolates from pepino(Solanum muricatum)[J]. Plant Dis, 2019, 103(7):1746-1756.
doi: 10.1094/PDIS-12-18-2164-RE pmid: 31082318 |
| [3] |
Wang N, Wang LY, Wang ZH, et al. Solanum muricatum ameliorates the symptoms of osteogenesis imperfecta in vivo[J]. J Food Sci, 2019, 84(6):1646-1650.
doi: 10.1111/1750-3841.14637 pmid: 31116433 |
| [4] | 岳恒. 香瓜茄多糖结构表征及免疫调节活性的研究[D]. 上海: 上海海洋大学, 2020. |
| Yue H. The structure characterization and immunomodulatory activity of polysaccharide from Solanum muricatum[D]. Shanghai: Shanghai Ocean University, 2020. | |
| [5] |
Zhao YQ, Zuo JH, Yuan SZ, et al. UV-C treatment maintains the sensory quality, antioxidant activity and flavor of pepino fruit during postharvest storage[J]. Foods, 2021, 10(12):2964.
doi: 10.3390/foods10122964 URL |
| [6] |
Pacheco J, Soler S, Figàs MR, et al. Screening of pepino(Solanum muricatum)and wild relatives against four major tomato diseases threatening its expansion in the Mediterranean region[J]. Ann Appl Biol, 2021, 179(3):288-301.
doi: 10.1111/aab.12698 URL |
| [7] |
Herraiz FJ, Villaño D, Plazas M, et al. Phenolic profile and biological activities of the pepino(Solanum muricatum)fruit and its wild relative S. caripense[J]. Int J Mol Sci, 2016, 17(3):394.
doi: 10.3390/ijms17030394 URL |
| [8] |
Hsu JY, Lin HH, Hsu CC, et al. Aqueous extract of pepino(Solanum muriactum Ait)leaves ameliorate lipid accumulation and oxidative stress in alcoholic fatty liver disease[J]. Nutrients, 2018, 10(7):931.
doi: 10.3390/nu10070931 URL |
| [9] |
Herraiz FJ, Blanca J, Ziarsolo P, et al. The first de novo transcriptome of pepino(Solanum muricatum):assembly, comprehensive analysis and comparison with the closely related species S. caripense, potato and tomato[J]. BMC Genomics, 2016, 17:321.
doi: 10.1186/s12864-016-2656-8 URL |
| [10] |
Yang SP, Zhu HD, Huang LP, et al. Transcriptome-wide and expression analysis of the NAC gene family in pepino(Solanum muricatum)during drought stress[J]. PeerJ, 2021, 9:e10966.
doi: 10.7717/peerj.10966 URL |
| [11] |
Su X, Wang BA, Geng XL, et al. A high-continuity and annotated tomato reference genome[J]. BMC Genomics, 2021, 22(1):898.
doi: 10.1186/s12864-021-08212-x pmid: 34911432 |
| [12] |
Kim S, Park J, Yeom SI, et al. New reference genome sequences of hot pepper reveal the massive evolution of plant disease-resistance genes by retroduplication[J]. Genome Biol, 2017, 18(1):210.
doi: 10.1186/s13059-017-1341-9 pmid: 29089032 |
| [13] |
Qin C, Yu CS, Shen YO, et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization[J]. Proc Natl Acad Sci USA, 2014, 111(14):5135-5140.
doi: 10.1073/pnas.1400975111 URL |
| [14] |
Kyriakidou M, Anglin NL, Ellis D, et al. Genome assembly of six polyploid potato genomes[J]. Sci Data, 2020, 7(1):88.
doi: 10.1038/s41597-020-0428-4 pmid: 32161269 |
| [15] |
Hirakawa H, Shirasawa K, Miyatake K, et al. Draft genome sequence of eggplant(Solanum melongena L.):the representative Solanum species indigenous to the old world[J]. DNA Res, 2014, 21(6):649-660.
doi: 10.1093/dnares/dsu027 pmid: 25233906 |
| [16] |
Sierro N, Battey JND, Ouadi S, et al. The tobacco genome sequence and its comparison with those of tomato and potato[J]. Nat Commun, 2014, 5:3833.
doi: 10.1038/ncomms4833 pmid: 24807620 |
| [17] |
Takei H, Shirasawa K, Kuwabara K, et al. De novo genome assembly of two tomato ancestors, Solanum pimpinellifolium and Solanum lycopersicum var. cerasiforme, by long-read sequencing[J]. DNA Res, 2021, 28(1):dsaa029.
doi: 10.1093/dnares/dsaa029 URL |
| [18] |
Bolger A, Scossa F, Bolger ME, et al. The genome of the stress-tolerant wild tomato species Solanum pennellii[J]. Nat Genet, 2014, 46(9):1034-1038.
doi: 10.1038/ng.3046 URL |
| [19] |
Zhou Q, Tang D, Huang W, et al. Haplotype-resolved genome analyses of a heterozygous diploid potato[J]. Nat Genet, 2020, 52(10):1018-1023.
doi: 10.1038/s41588-020-0699-x pmid: 32989320 |
| [20] |
Sun HQ, Jiao WB, Krause K, et al. Chromosome-scale and haplotype-resolved genome assembly of a tetraploid potato cultivar[J]. Nat Genet, 2022, 54(3):342-348.
doi: 10.1038/s41588-022-01015-0 pmid: 35241824 |
| [21] |
Rao SSP, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping[J]. Cell, 2014, 159(7):1665-1680.
doi: 10.1016/j.cell.2014.11.021 pmid: 25497547 |
| [22] |
Wang MC, Tong SF, Ma T, et al. Chromosome-level genome assembly of Sichuan pepper provides insights into apomixis, drought tolerance, and alkaloid biosynthesis[J]. Mol Ecol Resour, 2021, 21(7):2533-2545.
doi: 10.1111/1755-0998.13449 URL |
| [23] |
Yoon YJ, Venkatesh J, Lee JH, et al. Genome editing of eIF4E1 in tomato confers resistance to pepper mottle virus[J]. Front Plant Sci, 2020, 11:1098.
doi: 10.3389/fpls.2020.01098 pmid: 32849681 |
| [24] | 李宁, 王娟, 王柏柯, 等. 潘那利番茄SpCDPK4基因的克隆、原核表达及表达模式分析[J]. 植物生理学报, 2021, 57(3):672-680. |
| Li N, Wang J, Wang BK, et al. Cloning, prokaryotic expression and expression pattern analysis of SpCDPK4 gene in Solanum pennellii[J]. Plant Physiol J, 2021, 57(3):672-680. | |
| [25] |
Sato S, Tabata S, Hirakawa H, et al. The tomato genome sequence provides insights into fleshy fruit evolution[J]. Nature, 2012, 485(7400):635-641.
doi: 10.1038/nature11119 URL |
| [26] | 王娟, 王柏柯, 李宁, 等. 基因编辑技术在番茄育种中的应用进展[J]. 植物生理学报, 2020, 56(12):2606-2616. |
| Wang J, Wang BK, Li N, et al. Advances in the application of genome editing in tomato breeding[J]. Plant Physiol J, 2020, 56(12):2606-2616. | |
| [27] |
Salava H, Thula S, Mohan V, et al. Application of genome editing in tomato breeding:mechanisms, advances, and prospects[J]. Int J Mol Sci, 2021, 22(2):682.
doi: 10.3390/ijms22020682 URL |
| [28] |
Li RQ, Zhu HM, Ruan J, et al. De novo assembly of human genomes with massively parallel short read sequencing[J]. Genome Res, 2010, 20(2):265-272.
doi: 10.1101/gr.097261.109 URL |
| [29] |
Rozen S, Skaletsky H. Primer 3 on the WWW for general users and for biologist programmers[J]. Methods Mol Biol, 2000, 132:365-386.
pmid: 10547847 |
| [30] |
Flynn JM, Hubley R, Goubert C, et al. RepeatModeler2 for automated genomic discovery of transposable element families[J]. Proc Natl Acad Sci USA, 2020, 117(17):9451-9457.
doi: 10.1073/pnas.1921046117 URL |
| [31] |
Ou SJ, Jiang N. LTR_retriever:a highly accurate and sensitive program for identification of long terminal repeat retrotransposons[J]. Plant Physiol, 2018, 176(2):1410-1422.
doi: 10.1104/pp.17.01310 URL |
| [32] |
Bao ZR, Eddy SR. Automated de novo identification of repeat sequence families in sequenced genomes[J]. Genome Res, 2002, 12(8):1269-1276.
doi: 10.1101/gr.88502 URL |
| [33] | Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes[J]. Bioinformatics, 2005, 21(Suppl 1):i351-i358. |
| [34] |
Jurka J, Kapitonov VV, Pavlicek A, et al. Repbase update, a database of eukaryotic repetitive elements[J]. Cytogenet Genome Res, 2005, 110(1/2/3/4):462-467.
doi: 10.1159/000084979 URL |
| [35] |
Neumann P, Novák P, Hoštáková N, et al. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification[J]. Mob DNA, 2019, 10:1.
doi: 10.1186/s13100-018-0144-1 URL |
| [36] | Wheeler TJ, Clements J, Eddy SR, et al. Dfam:a database of repetitive DNA based on profile hidden Markov models[J]. Nucleic Acids Res, 2013, 41(Database issue):D70-D82. |
| [37] | Chen NS. Using RepeatMasker to identify repetitive elements in genomic sequences[J]. Curr Protoc Bioinform, 2004, 5(1):4. 10.1-4. 10. 14. |
| [38] |
Korf I. Gene finding in novel genomes[J]. BMC Bioinformatics, 2004, 5:59.
pmid: 15144565 |
| [39] |
Grabherr MG, Haas BJ, Yassour M, et al. Trinity:reconstructing a full-length transcriptome without a genome from RNA-Seq data[J]. Nat Biotechnol, 2011, 29(7):644-652.
doi: 10.1038/nbt.1883 pmid: 21572440 |
| [40] |
Haas BJ, Salzberg SL, Zhu W, et al. Automated eukaryotic gene structure annotation using evidence modeler and the program to assemble spliced alignments[J]. Genome Biol, 2008, 9(1):R7.
doi: 10.1186/gb-2008-9-1-r7 URL |
| [41] | Griffiths-Jones S, Moxon S, Marshall M, et al. Rfam:annotating non-coding RNAs in complete genomes[J]. Nucleic Acids Res, 2005, 33(Database issue):D121-D124. |
| [42] |
Birney E, Clamp M, Durbin R. GeneWise and genomewise[J]. Genome Res, 2004, 14(5):988-995.
pmid: 15123596 |
| [43] |
Hu Y, Chen JD, Fang L, et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton[J]. Nat Genet, 2019, 51(4):739-748.
doi: 10.1038/s41588-019-0371-5 URL |
| [44] |
Gao ZY, Li ZH, Lin DL, et al. Chromosome-scale genome assembly of the resurrection plant Acanthochlamys bracteata(Velloziaceae)[J]. Genome Biol Evol, 2021, 13(8):evab147.
doi: 10.1093/gbe/evab147 URL |
| [45] |
Rabanus-Wallace MT, Hackauf B, Mascher M, et al. Chromosome-scale genome assembly provides insights into rye biology, evolution and agronomic potential[J]. Nat Genet, 2021, 53(4):564-573.
doi: 10.1038/s41588-021-00807-0 pmid: 33737754 |
| [46] |
Kang MH, Wu HL, Yang Q, et al. A chromosome-scale genome assembly of Isatis indigotica, an important medicinal plant used in traditional Chinese medicine:an Isatis genome[J]. Hortic Res, 2020, 7:18.
doi: 10.1038/s41438-020-0240-5 URL |
| [47] | 李育先. 茄科作物基因组的多重序列比对与进化研究[D]. 唐山: 华北理工大学, 2016. |
| Li YX. Genome multiple alignment and evolutionary studies of Solanaceae lineages[D]. Tangshan: North China University of Science and Technology, 2016. | |
| [48] |
Cao YL, Li YL, Fan YF, et al. Wolfberry genomes and the evolution of Lycium(Solanaceae)[J]. Commun Biol, 2021, 4(1):671.
doi: 10.1038/s42003-021-02152-8 URL |
| [49] | 苑克俊, 葛福荣, 牛庆霖. 杏基因组全新组装及杏的进化分析[J]. 植物生理学报, 2020, 56(10):2187-2200. |
| Yuan KJ, Ge FR, Niu QL. De novo apricot(Prunus armeniaca)genome assembly and evolutionary analysis[J]. Plant Physiol J, 2020, 56(10):2187-2200. |
| [1] | 王腾辉, 葛雯冬, 罗雅方, 范震宇, 王玉书. 基于极端混合池(BSA)全基因组重测序的羽衣甘蓝白色叶基因定位[J]. 生物技术通报, 2023, 39(9): 176-182. |
| [2] | 李雪琪, 张素杰, 于曼, 黄金光, 周焕斌. 基于CRISPR/CasX介导的水稻基因组编辑技术的建立[J]. 生物技术通报, 2023, 39(9): 40-48. |
| [3] | 方澜, 黎妍妍, 江健伟, 成胜, 孙正祥, 周燚. 盘龙参内生真菌胞内细菌7-2H的分离鉴定和促生特性研究[J]. 生物技术通报, 2023, 39(8): 272-282. |
| [4] | 饶紫环, 谢志雄. 一株Olivibacter jilunii 纤维素降解菌株的分离鉴定与降解能力分析[J]. 生物技术通报, 2023, 39(8): 283-290. |
| [5] | 郭少华, 毛会丽, 刘征权, 付美媛, 赵平原, 马文博, 李旭东, 关建义. 一株鱼源致病性嗜水气单胞菌XDMG的全基因组测序及比较基因组分析[J]. 生物技术通报, 2023, 39(8): 291-306. |
| [6] | 杜冬冬, 钱晶, 李思琪, 刘雯菲, 魏向利, 刘长勇, 罗瑞峰, 康立超. 单核细胞增生李斯特菌LMXJ15全基因组测序及分析[J]. 生物技术通报, 2023, 39(7): 298-306. |
| [7] | 李雨真, 梅天秀, 李治文, 王淇, 李俊, 邹岳, 赵心清. 红酵母基因组和代谢工程改造研究进展[J]. 生物技术通报, 2023, 39(7): 67-79. |
| [8] | 尹明华, 余锾媛, 肖心怡, 王玉婷. 江西铅山红芽芋叶绿体基因组特征及系统发育分析[J]. 生物技术通报, 2023, 39(6): 233-247. |
| [9] | 张路阳, 韩文龙, 徐晓雯, 姚健, 李芳芳, 田效园, 张智强. 烟草TCP基因家族的鉴定及表达分析[J]. 生物技术通报, 2023, 39(6): 248-258. |
| [10] | 赖瑞联, 冯新, 高敏霞, 路喻丹, 刘晓驰, 吴如健, 陈义挺. 猕猴桃过氧化氢酶基因家族全基因组鉴定与表达分析[J]. 生物技术通报, 2023, 39(4): 136-147. |
| [11] | 周晓杰, 杨思琪, 张译文, 徐佳琪, 杨晟. CRISPR相关转座酶及其细菌基因组编辑应用[J]. 生物技术通报, 2023, 39(4): 49-58. |
| [12] | 肖小军, 陈明, 韩德鹏, 余跑兰, 郑伟, 肖国滨, 周庆红, 周会汶. 甘蓝型油菜每角果粒数全基因组关联分析[J]. 生物技术通报, 2023, 39(3): 143-151. |
| [13] | 张志霞, 李天培, 曾虹, 朱稀贤, 杨天雄, 马斯楠, 黄磊. 冰冷杆菌PG-2的基因组测序及生物信息学分析[J]. 生物技术通报, 2023, 39(3): 290-300. |
| [14] | 汪格格, 邱诗蕊, 张琳晗, 杨国伟, 徐小云, 汪爱羚, 曾淑华, 刘雅洁. 异源三倍体普通烟草(SST)减数分裂期的分子细胞学研究[J]. 生物技术通报, 2023, 39(2): 183-192. |
| [15] | 和梦颖, 刘文彬, 林震鸣, 黎尔彤, 汪洁, 金小宝. 一株抗革兰阳性菌的戈登氏菌WA4-43全基因组测序与分析[J]. 生物技术通报, 2023, 39(2): 232-242. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||