







生物技术通报 ›› 2024, Vol. 40 ›› Issue (10): 98-107.doi: 10.13560/j.cnki.biotech.bull.1985.2024-0788
高云云1( ), 杨海飞1,2, 吕虎杰1, 刘永鑫1()
), 杨海飞1,2, 吕虎杰1, 刘永鑫1()
收稿日期:2024-08-16
出版日期:2024-10-26
发布日期:2024-11-20
通讯作者:
刘永鑫,男,博士,研究员,研究方向:微生物组方法开发、功能挖掘与科学传播;E-mail: liuyongxin@caas.cn作者简介:高云云,女,博士,博士后,研究方向:宏基因组数据分析的软件评测、流程搭建与优化;E-mail: gaoyunyun@caas.cn
基金资助:
GAO Yun-yun1(), YANG Hai-fei1,2, LYU Hu-jie1, LIU Yong-xin1()
Received:2024-08-16
Published:2024-10-26
Online:2024-11-20
摘要:
微生物是生命科学研究中不可或缺的重要资源,其研究对推动科学进步、促进人类健康和改善环境质量等具有重要意义。随着二、三代高通量测序技术的迅猛发展,我们对微生物世界的认知得到了极大提升,在面对大量的微生物组数据时,选择适当的分析方法以实现快速、准确的信息挖掘显得尤为关键。本文对近年来微生物组研究的进展进行了系统的回顾与分析,着重更新了扩增子、培养组和宏基因组等二代短读序宏基因组数据的分析工具,纳入了三代长读序宏基因数据的处理方案,并提出了标准化数据分析流程的必要性。此外,本文结合解析微生物组的实例案例,侧重介绍其在植物与根系微生物互作、微生物多样性等方面的应用实例,探讨了不同方法在微生物组构成、结构和功能分析中的优劣,展示了宏基因组数据挖掘在应用方面的潜力,以期拓宽宏基因组数据挖掘的研究思路。最后,本文指出了当前微生物组研究中的不足和面临的挑战,并展望了未来微生物组研究技术的标准化与流程化方面的发展趋势,以期加速微生物组的功能与应用的研究进程。
高云云, 杨海飞, 吕虎杰, 刘永鑫. 微生物组分析方法与功能挖掘[J]. 生物技术通报, 2024, 40(10): 98-107.
GAO Yun-yun, YANG Hai-fei, LYU Hu-jie, LIU Yong-xin. Analytical Approaches and Functional Insights for Microbiome Studies[J]. Biotechnology Bulletin, 2024, 40(10): 98-107.

图1 微生物组研究的发展与应用 A:微生物组研究的应用场景;B:重大的技术和方法发展推动微生物组的研究;C:微生物组研究热点的发展趋势
Fig. 1 Development and application of microbiome study A: Application scenarios of microbiome study. B: Significant technological and methodological developments driving microbiome study. C: Trends in key areas of microbiome study
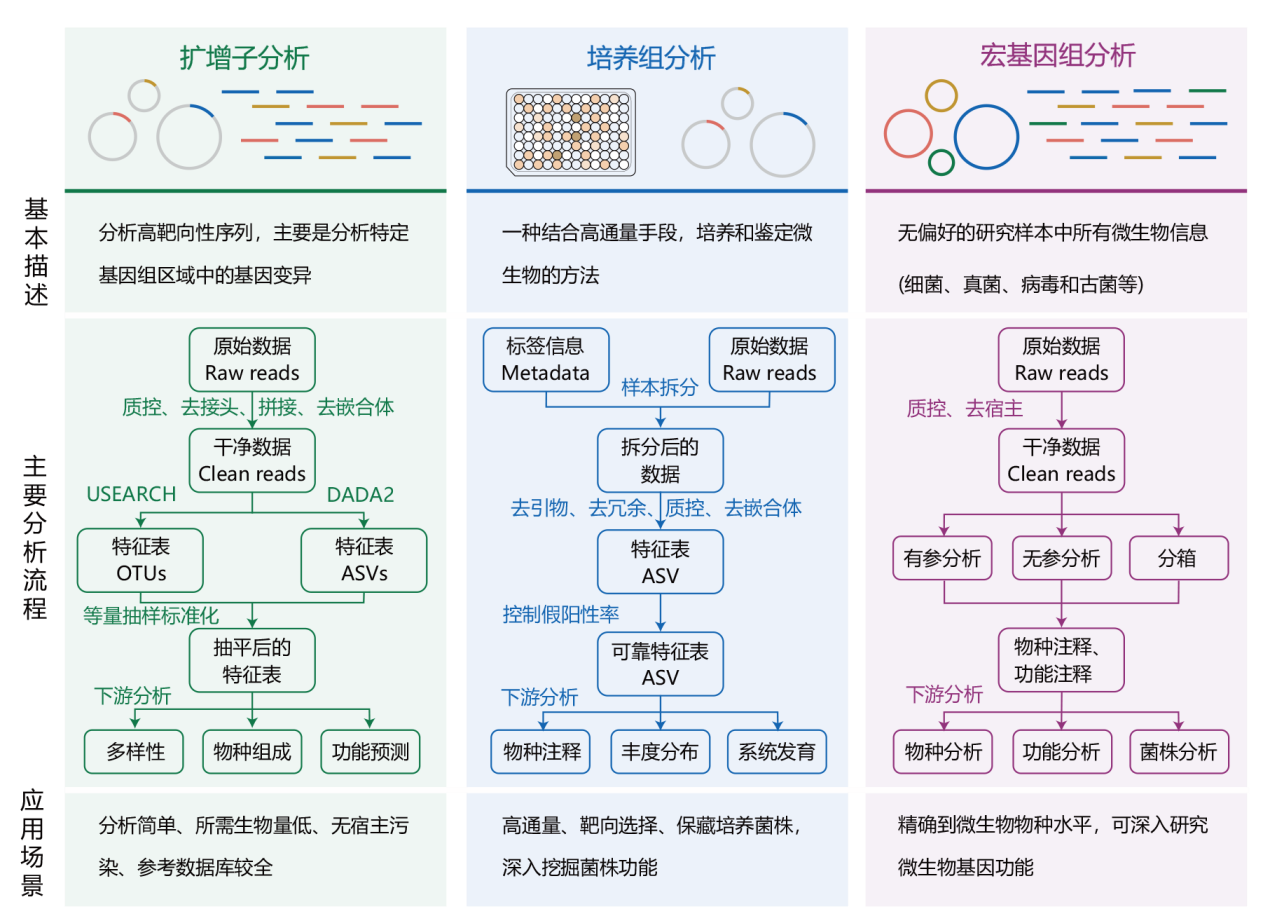
图2 基于二代高通量测序技术的分析流程和应用方案
Fig. 2 Analysis workflow and applications based on second-generation high-throughput sequencing technology

图3 基于宏基因组分析探讨植物与根际微生物的互作对植物营养吸收影响的案例 A:提出科学假设;根据籼稻氮肥利用效率高于粳稻这一科学现象提出,水稻和根际微生物组如何互作影响氮肥的利用效率这一科学假设;B:开展实践方案;基于扩增子、宏基因组、培养组分析,发现籼粳稻根系微生物组差异分析、根系微生物组差异受NRT1.1B基因调控、水稻根系微生物培养和合成菌群改变水稻氮吸收实验的实践方案;C:获取理论验证;特定根系微生物会参与有机氮矿化的过程促进水稻的氮元素吸收,且该现象与硝酸盐转运蛋白基因NRT1.1B在籼粳稻之间的自然变异相关联
Fig. 3 Case study on the effects of plants-rhizosphere microorganism interaction on plant nutrient uptake based on metagenomic analysis A: Develop scientific hypotheses. Based on the observation that nitrogen use efficiency of indica rice is higher than that of japonica rice, the hypothesis that rice and the rhizosphere microbiome interact to influence nitrogen use efficiency is proposed. B: Implement practical programs. Utilizing amplification, metagenomic analysis, and culture group analysis, the differences in root microbiomes between indica and japonica rice are examined. Additionally, the role of the NRT1.1B gene in regulating these differences is investigated, and experiments are conducted on the cultivation of rice root microbiomes and synthetic communities to assess changes in nitrogen absorption. C: Theoretical validation. Specific root microbiomes participate in the process of organic nitrogen mineralization, thereby enhancing nitrogen uptake in rice. This phenomenon is linked to the natural variation of the nitrate transporter gene NRT1.1B between indica and japonica rice

图4 基于三代纳米孔技术的分析流程和应用方案
Fig. 4 Analysis workflow and applications based on third-generation nanopore technology
| [1] | Kumari P, Prakash P, Yadav S, et al. Microbiome analysis: an emerging forensic investigative tool[J]. Forensic Sci Int, 2022, 340: 111462. |
| [2] |
Berg G, Rybakova D, Fischer D, et al. Microbiome definition re-visited: old concepts and new challenges[J]. Microbiome, 2020, 8(1): 103.
doi: 10.1186/s40168-020-00875-0 pmid: 32605663 |
| [3] | 刘双江, 施文元, 赵国屏. 中国微生物组计划: 机遇与挑战[J]. 中国科学院院刊, 2017, 32(3): 241-250. |
| Liu SJ, Shi WY, Zhao GP. China microbiome initiative: opportunity and challenges[J]. Bull Chin Acad Sci, 2017, 32(3): 241-250. | |
| [4] | 刘永鑫, 秦媛, 郭晓璇, 等. 微生物组数据分析方法与应用[J]. 遗传, 2019, 41(9): 845-862. |
| Liu YX, Qin Y, Guo XX, et al. Methods and applications for microbiome data analysis[J]. Hereditas, 2019, 41(9): 845-862. | |
| [5] |
Enge M, Arda HE, Mignardi M, et al. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns[J]. Cell, 2017, 171(2): 321-330.e14.
doi: S0092-8674(17)31053-X pmid: 28965763 |
| [6] |
Earl JP, Adappa ND, Krol J, et al. Species-level bacterial community profiling of the healthy sinonasal microbiome using Pacific Biosciences sequencing of full-length 16S rRNA genes[J]. Microbiome, 2018, 6(1): 190.
doi: 10.1186/s40168-018-0569-2 pmid: 30352611 |
| [7] |
Karst SM, Ziels RM, Kirkegaard RH, et al. High-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or PacBio sequencing[J]. Nat Methods, 2021, 18(2): 165-169.
doi: 10.1038/s41592-020-01041-y pmid: 33432244 |
| [8] |
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads[J]. Nat Methods, 2013, 10(10): 996-998.
doi: 10.1038/nmeth.2604 pmid: 23955772 |
| [9] |
Callahan BJ, McMurdie PJ, Rosen MJ, et al. DADA2: High-resolution sample inference from Illumina amplicon data[J]. Nat Methods, 2016, 13(7): 581-583.
doi: 10.1038/nmeth.3869 pmid: 27214047 |
| [10] | Liu YX, Qin Y, Chen T, et al. A practical guide to amplicon and metagenomic analysis of microbiome data[J]. Protein Cell, 2021, 12(5): 315-330. |
| [11] |
Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nat Methods, 2010, 7(5): 335-336.
doi: 10.1038/nmeth.f.303 pmid: 20383131 |
| [12] |
Bolyen E, Rideout JR, Dillon MR, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2[J]. Nat Biotechnol, 2019, 37(8): 852-857.
doi: 10.1038/s41587-019-0209-9 pmid: 31341288 |
| [13] | Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities[J]. Appl Environ Microbiol, 2009, 75(23): 7537-7541. |
| [14] | Liu YX, Chen L, Ma TF, et al. EasyAmplicon: an easy-to-use, open-source, reproducible, and community-based pipeline for amplicon data analysis in microbiome research[J]. iMeta, 2023, 2(1): e83. |
| [15] | Gao YY, Zhang GX, Jiang SY, et al. Wekemo Bioincloud: a user-friendly platform for meta-omics data analyses[J]. iMeta, 2024, 3(1): e175. |
| [16] |
Martiny AC. High proportions of bacteria are culturable across major biomes[J]. ISME J, 2019, 13(8): 2125-2128.
doi: 10.1038/s41396-019-0410-3 pmid: 30952994 |
| [17] |
Zhang JY, Liu YX, Guo XX, et al. High-throughput cultivation and identification of bacteria from the plant root microbiota[J]. Nat Protoc, 2021, 16(2): 988-1012.
doi: 10.1038/s41596-020-00444-7 pmid: 33442053 |
| [18] | Xia H, Zhang Z, Luo C, et al. MultiPrime: a reliable and efficient tool for targeted next-generation sequencing[J]. iMeta, 2023, 2(4): e143. |
| [19] |
玉霞, 赵飞燕, 孙志宏. 肠道中有益菌培养组学的研究进展[J]. 食品科学, 2023, 44(23): 365-371.
doi: 10.7506/spkx1002-6630-20221210-103 |
| Yu X, Zhao FY, Sun ZH. Progress in culturemics research on beneficial intestinal bacteria[J]. Food Sci, 2023, 44(23): 365-371. | |
| [20] | 王慧, 鞠峰. 宏基因组学在环境微生物组研究中的应用与展望[J]. 微生物学通报, 2024, 51(6): 1814-1833. |
| Wang H, Ju F. Applications and perspectives of metagenomics in environmental microbiome research[J]. Microbiol China, 2024, 51(6): 1814-1833. | |
| [21] |
Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2[J]. Genome Biol, 2019, 20(1): 257.
doi: 10.1186/s13059-019-1891-0 pmid: 31779668 |
| [22] |
Franzosa EA, McIver LJ, Rahnavard G, et al. Species-level functional profiling of metagenomes and metatranscriptomes[J]. Nat Methods, 2018, 15(11): 962-968.
doi: 10.1038/s41592-018-0176-y pmid: 30377376 |
| [23] | Beghini F, McIver LJ, Blanco-Míguez A, et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3[J]. eLife, 2021, 10: e65088. |
| [24] |
Zhang JY, Liu YX, Zhang N, et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice[J]. Nat Biotechnol, 2019, 37(6): 676-684.
doi: 10.1038/s41587-019-0104-4 pmid: 31036930 |
| [25] |
Callahan BJ, Grinevich D, Thakur S, et al. Ultra-accurate microbial amplicon sequencing with synthetic long reads[J]. Microbiome, 2021, 9(1): 130.
doi: 10.1186/s40168-021-01072-3 pmid: 34090540 |
| [26] | Wick RR, Judd LM, Gorrie CL, et al. Completing bacterial genome assemblies with multiplex MinION sequencing[J]. Microb Genom, 2017, 3(10): e000132. |
| [27] |
Johnson JS, Spakowicz DJ, Hong BY, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis[J]. Nat Commun, 2019, 10(1): 5029.
doi: 10.1038/s41467-019-13036-1 pmid: 31695033 |
| [28] | Callahan BJ, Wong J, Heiner C, et al. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution[J]. Nucleic Acids Res, 2019, 47(18): e103. |
| [29] | Moon J, Kim N, Kim TJ, et al. Rapid diagnosis of bacterial meningitis by nanopore 16S amplicon sequencing: a pilot study[J]. Int J Med Microbiol, 2019, 309(6): 151338. |
| [30] | Kai S, Matsuo Y, Nakagawa S, et al. Rapid bacterial identification by direct PCR amplification of 16S rRNA genes using the MinIONTM nanopore sequencer[J]. FEBS Open Bio, 2019, 9(3): 548-557. |
| [31] |
Ohta A, Nishi K, Hirota K, et al. Using nanopore sequencing to identify fungi from clinical samples with high phylogenetic resolution[J]. Sci Rep, 2023, 13(1): 9785.
doi: 10.1038/s41598-023-37016-0 pmid: 37328565 |
| [32] |
Benítez-Páez A, Portune KJ, Sanz Y. Species-level resolution of 16S rRNA gene amplicons sequenced through the MinIONTM portable nanopore sequencer[J]. Gigascience, 2016, 5: 4.
doi: 10.1186/s13742-016-0111-z pmid: 26823973 |
| [33] |
Rang FJ, Kloosterman WP, de Ridder J. From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy[J]. Genome Biol, 2018, 19(1): 90.
doi: 10.1186/s13059-018-1462-9 pmid: 30005597 |
| [34] |
Wang YH, Zhao Y, Bollas A, et al. Nanopore sequencing technology, bioinformatics and applications[J]. Nat Biotechnol, 2021, 39(11): 1348-1365.
doi: 10.1038/s41587-021-01108-x pmid: 34750572 |
| [35] |
Lorig-Roach R, Meredith M, Monlong J, et al. Phased nanopore assembly with Shasta and modular graph phasing with GFAse[J]. Genome Res, 2024, 34(3): 454-468.
doi: 10.1101/gr.278268.123 pmid: 38627094 |
| [36] | Wang Z, Qin L, Liu J, et al. Forensic nanopore sequencing of microhaplotype markers using QitanTech's QNome[J]. Forensic Sci Int Genet, 2022, 57: 102657. |
| [37] | Ciuffreda L, Rodríguez-Pérez H, Flores C. Nanopore sequencing and its application to the study of microbial communities[J]. Comput Struct Biotechnol J, 2021, 19: 1497-1511. |
| [38] | Overholt WA, Hölzer M, Geesink P, et al. Inclusion of Oxford Nanopore long reads improves all microbial and viral metagenome-assembled genomes from a complex aquifer system[J]. Environ Microbiol, 2020, 22(9): 4000-4013. |
| [39] |
Kolmogorov M, Bickhart DM, Behsaz B, et al. metaFlye: scalable long-read metagenome assembly using repeat graphs[J]. Nat Methods, 2020, 17(11): 1103-1110.
doi: 10.1038/s41592-020-00971-x pmid: 33020656 |
| [40] | Koren S, Walenz BP, Berlin K, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation[J]. Genome Res, 2017, 27(5): 722-736. |
| [41] |
Ruan J, Li H. Fast and accurate long-read assembly with wtdbg2[J]. Nat Methods, 2020, 17(2): 155-158.
doi: 10.1038/s41592-019-0669-3 pmid: 31819265 |
| [42] |
Hu J, Wang Z, Sun ZY, et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads[J]. Genome Biol, 2024, 25(1): 107.
doi: 10.1186/s13059-024-03252-4 pmid: 38671502 |
| [43] |
Bertrand D, Shaw J, Kalathiyappan M, et al. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes[J]. Nat Biotechnol, 2019, 37(8): 937-944.
doi: 10.1038/s41587-019-0191-2 pmid: 31359005 |
| [44] |
Nurk S, Meleshko D, Korobeynikov A, et al. metaSPAdes: a new versatile metagenomic assembler[J]. Genome Res, 2017, 27(5): 824-834.
doi: 10.1101/gr.213959.116 pmid: 28298430 |
| [45] | Wick RR, Judd LM, Gorrie CL, et al. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads[J]. PLoS Comput Biol, 2017, 13(6): e1005595. |
| [46] |
Zhang HW, Jain C, Aluru S. A comprehensive evaluation of long read error correction methods[J]. BMC Genomics, 2020, 21(Suppl 6): 889.
doi: 10.1186/s12864-020-07227-0 pmid: 33349243 |
| [47] |
Fu SH, Wang AQ, Au KF. A comparative evaluation of hybrid error correction methods for error-prone long reads[J]. Genome Biol, 2019, 20(1): 26.
doi: 10.1186/s13059-018-1605-z pmid: 30717772 |
| [48] | Agustinho DP, Fu YL, Menon VK, et al. Unveiling microbial diversity: harnessing long-read sequencing technology[J]. Nat Methods, 2024, 21(6): 954-966. |
| [49] |
Sedlazeck FJ, Rescheneder P, Smolka M, et al. Accurate detection of complex structural variations using single-molecule sequencing[J]. Nat Methods, 2018, 15(6): 461-468.
doi: 10.1038/s41592-018-0001-7 pmid: 29713083 |
| [50] | Peng K, Liu YX, Sun XR, et al. Large-scale bacterial genomic and metagenomic analysis reveals Pseudomonas aeruginosa as potential ancestral source of tigecycline resistance gene cluster tmexCD-toprJ[J]. Microbiol Res, 2024, 285: 127747. |
| [51] |
Isidro J, Borges V, Pinto M, et al. Phylogenomic characterization and signs of microevolution in the 2022 multi-country outbreak of monkeypox virus[J]. Nat Med, 2022, 28(8): 1569-1572.
doi: 10.1038/s41591-022-01907-y pmid: 35750157 |
| [52] |
Gao YY, Li DY, Liu YX. Microbiome research outlook: past, present, and future[J]. Protein Cell, 2023, 14(10): 709-712.
doi: 10.1093/procel/pwad031 |
| [1] | 王昕璐, 王蒙, 翟文磊. 脂质组学在毒理学研究中的应用[J]. 生物技术通报, 2023, 39(3): 69-80. |
| [2] | 李凯航, 王浩臣, 程可心, 杨艳, 金一, 何晓青. 全基因组关联分析研究植物与微生物组的互作遗传机制[J]. 生物技术通报, 2023, 39(2): 24-34. |
| [3] | 张岩峰, 丁燕玲, 马应, 周小南, 杨朝云, 史远刚, 康晓龙. 肉牛剩余采食量相关瘤胃及粪便微生物特征比较分析[J]. 生物技术通报, 2023, 39(1): 295-304. |
| [4] | 鲁兆祥, 王夕冉, 连新磊, 廖晓萍, 刘雅红, 孙坚. 基于功能宏基因组学挖掘抗生素耐药基因研究进展[J]. 生物技术通报, 2022, 38(9): 17-27. |
| [5] | 钟辉, 刘亚军, 王滨花, 和梦洁, 吴兰. 分析方法对细菌群落16S rRNA基因扩增测序分析结果的影响[J]. 生物技术通报, 2022, 38(6): 81-92. |
| [6] | 王宁, 李蕙秀, 李季, 丁国春. 堆肥调控作物根际微生物组抑制植物病害的研究进展[J]. 生物技术通报, 2022, 38(5): 4-12. |
| [7] | 张雨函, 范熠, 李婷婷, 庞爽, 刘为, 白可喻, 张西美. 基于宏基因组测序的植物叶表微生物富集及DNA提取方法[J]. 生物技术通报, 2022, 38(3): 256-263. |
| [8] | 袁源, 黄海辰, 李琳, 刘国辉, 傅俊生, 吴小平. 石灰对灵芝覆土连作障碍的防控作用及其微生物群落分析[J]. 生物技术通报, 2021, 37(4): 70-84. |
| [9] | 李叶青, 景张牧, 江皓, 徐泉, 周红军, 冯璐. 微生物组学及其在厌氧消化中的研究进展[J]. 生物技术通报, 2021, 37(1): 90-101. |
| [10] | 马涛, 陆唯, 李松励, 樊霞. 畜禽微生物耐药组研究进展[J]. 生物技术通报, 2021, 37(1): 113-122. |
| [11] | 张立兴, 王丽娜, 康广博, 黄鹤. 多组学分析在炎症性肠病中的应用与研究进展[J]. 生物技术通报, 2021, 37(1): 155-167. |
| [12] | 陆玉芳, 施卫明. 根际化学信号物质与土壤养分转化[J]. 生物技术通报, 2020, 36(9): 14-24. |
| [13] | 孙雨, 常晶晶, 田春杰. 作物根际微生物组重组构建技术体系探讨[J]. 生物技术通报, 2020, 36(9): 25-30. |
| [14] | 汪盼盼, 杨野, 刘迪秋, 崔秀明, 刘源. 宏基因组学在植物病害研究中的应用[J]. 生物技术通报, 2020, 36(12): 146-154. |
| [15] | 刘纪爱, 束爱萍, 刘光荣, 李祖章, 刘增兵, 高峥. 施肥影响土壤性状和微生物组的研究进展[J]. 生物技术通报, 2019, 35(9): 21-28. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||