







生物技术通报 ›› 2025, Vol. 41 ›› Issue (12): 177-189.doi: 10.13560/j.cnki.biotech.bull.1985.2025-0451
许聪聪1,2( ), 郑美1, 李翠1, 赵春桥1, 何玮2, 侯新村1(), 郭强1()
), 郑美1, 李翠1, 赵春桥1, 何玮2, 侯新村1(), 郭强1()
收稿日期:2025-05-01
出版日期:2025-12-26
发布日期:2026-01-06
通讯作者:
侯新村,男,副研究员,研究方向 :饲草抗逆遗传育种;E-mail: houxincun@baafs.net.cn作者简介:许聪聪,女,硕士研究生,研究方向 :植物逆境生理与生物育种;E-mail: 202332680@stumail.nwu.edu.cn
基金资助:
XU Cong-cong1,2(), ZHENG Mei1, LI Cui1, ZHAO Chun-qiao1, HE Wei2, HOU Xin-cun1(), GUO Qiang1()
Received:2025-05-01
Published:2025-12-26
Online:2026-01-06
摘要:
目的 通过分析盐处理下花花柴转录组数据,挖掘与之耐盐性相关的特异性基因模块,旨在为深入理解花花柴耐盐适应性分子机制提供理论依据。 方法 以不同盐浓度处理的花花柴叶片为研究对象,基于转录组和加权基因共表达网络分析(Weighted Gene Co-expression Network Analysis, WGCNA)鉴定与其耐盐相关的特异性基因模块,以此筛选关键基因。 结果 转录组测序产生了103.66 GB的数据,组装得到66 823个Unigene。其中,45.15%(30 171个)在6个公共蛋白质数据库中获得10 243个差异表达基因(DEGs)注释。同时,通过WGCNA分析,从其显著上调的DEGs中识别出10个共表达模块。值得注意的是,Brown模块、Pink模块、Yellow模块分别与400、300、200 mmol/L NaCl处理呈正相关。进一步,对这些特异性模块内的基因进行KEGG分析,发现它们主要在次生代谢物合成、植物激素信号转导和MAPK信号通路等生物学过程中显著富集。因此,基于模块内基因的连接度和注释信息,筛选出PILS6、REM4.1、DOF21、MAPKKK18、GATA8-like、SAUR76、ABH、CIPK6、DIR22、4CL2等核心基因。由此表明,这些基因可能在参与花花柴耐盐适应性中起重要的作用。 结论 通过转录组和WGCNA分析,筛选出与花花柴耐盐相关的核心基因和特异性模块。为花花柴耐盐基因资源挖掘与利用奠定基础。
许聪聪, 郑美, 李翠, 赵春桥, 何玮, 侯新村, 郭强. 利用WGCNA 筛选鉴定花花柴耐盐核心基因[J]. 生物技术通报, 2025, 41(12): 177-189.
XU Cong-cong, ZHENG Mei, LI Cui, ZHAO Chun-qiao, HE Wei, HOU Xin-cun, GUO Qiang. Screening and Identification of Salt-tolerant Hub Genes in Karelinia caspia Using WGCNA[J]. Biotechnology Bulletin, 2025, 41(12): 177-189.
模块 Module | 基因编号 Gene ID | 基因名称 Gene name | 正向引物 Forward primer (5'-3') | 反向引物 Reverse primer (5'-3') |
|---|---|---|---|---|
表1 核心基因定量引物表
Table 1 Primers of hub genes
模块 Module | 基因编号 Gene ID | 基因名称 Gene name | 正向引物 Forward primer (5'-3') | 反向引物 Reverse primer (5'-3') |
|---|---|---|---|---|
模块 Module | 核心基因 Hub gene ID | 基因名称 Gene name | 基因描述 Gene description |
|---|---|---|---|
| RNase H | |||
| PILS6 | |||
| - | |||
| - | |||
| GS | |||
| REM4.1 | |||
| - | |||
| - | |||
| BAM9 | |||
| DOF21 | |||
| - | |||
| MAPKKK18 | |||
| GATA8 | |||
| SAUR76 | |||
| RD19D | |||
| - | |||
| GRD | |||
| - | |||
| - | |||
| - | |||
| Rubisco | |||
| ABH | |||
| SULTR3.1 | |||
| CIPK6 | |||
| PE2 | |||
| KIN | |||
| - | |||
| DIR22 | |||
| 4CL2 | |||
| HIPPs47 |
表2 特异模块中花花柴耐盐相关的核心基因及功能注释
Table 2 Hub genes related to salt-tolerance in Karelinia caspia and functional annotations in specific modules
模块 Module | 核心基因 Hub gene ID | 基因名称 Gene name | 基因描述 Gene description |
|---|---|---|---|
| RNase H | |||
| PILS6 | |||
| - | |||
| - | |||
| GS | |||
| REM4.1 | |||
| - | |||
| - | |||
| BAM9 | |||
| DOF21 | |||
| - | |||
| MAPKKK18 | |||
| GATA8 | |||
| SAUR76 | |||
| RD19D | |||
| - | |||
| GRD | |||
| - | |||
| - | |||
| - | |||
| Rubisco | |||
| ABH | |||
| SULTR3.1 | |||
| CIPK6 | |||
| PE2 | |||
| KIN | |||
| - | |||
| DIR22 | |||
| 4CL2 | |||
| HIPPs47 |
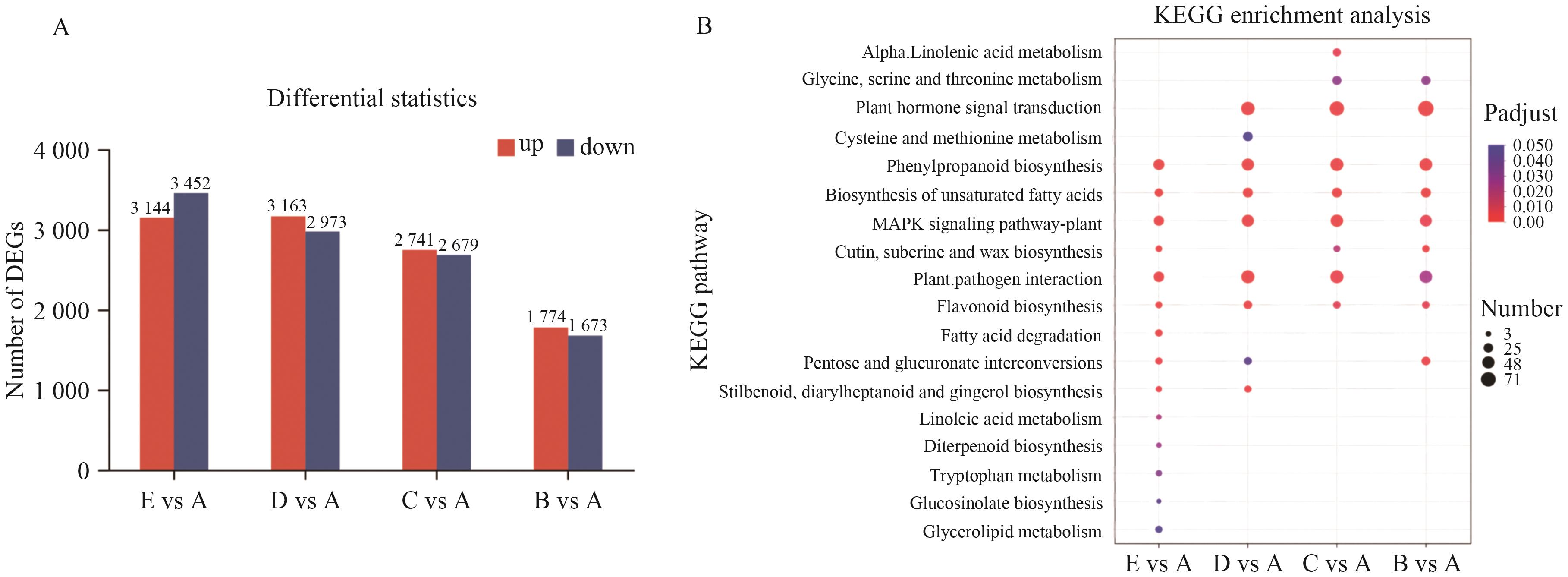
图1 各组差异表达基因统计(A)及功能富集图(B)A: CK; B: 100 mmol/L NaCl; C: 200 mmol/L NaCl; D: 300 mmol/L NaCl; E: 400 mmol/L NaCl. The same below
Fig. 1 Expression level differences (A) and KEGG enrichment analysis (B) of differentially expressed genes
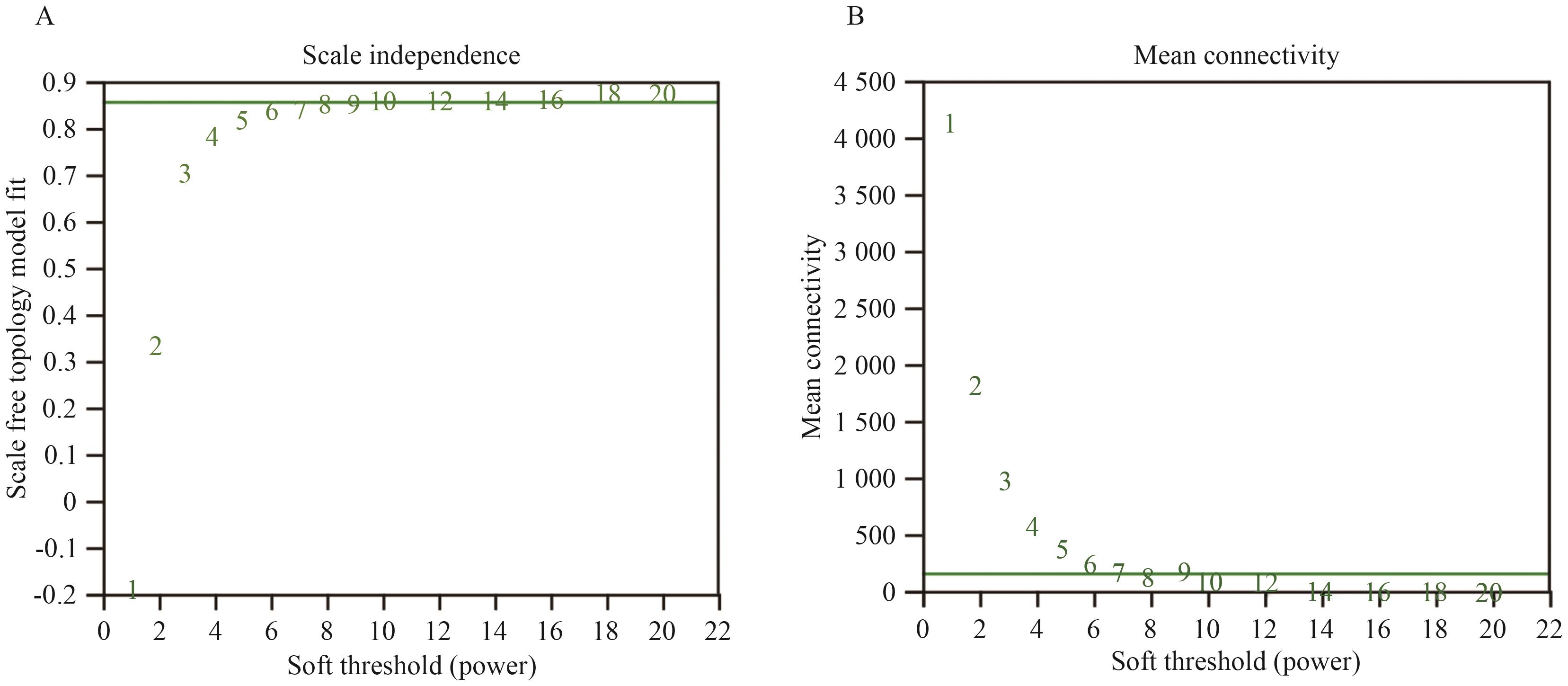
图2 软阈值Power值的确定A:无尺度网络模型图;B:网络平均连接度
Fig. 2 Determination of soft threshold Power valueA: Scale-free network model diagram. B: Network average connectivity
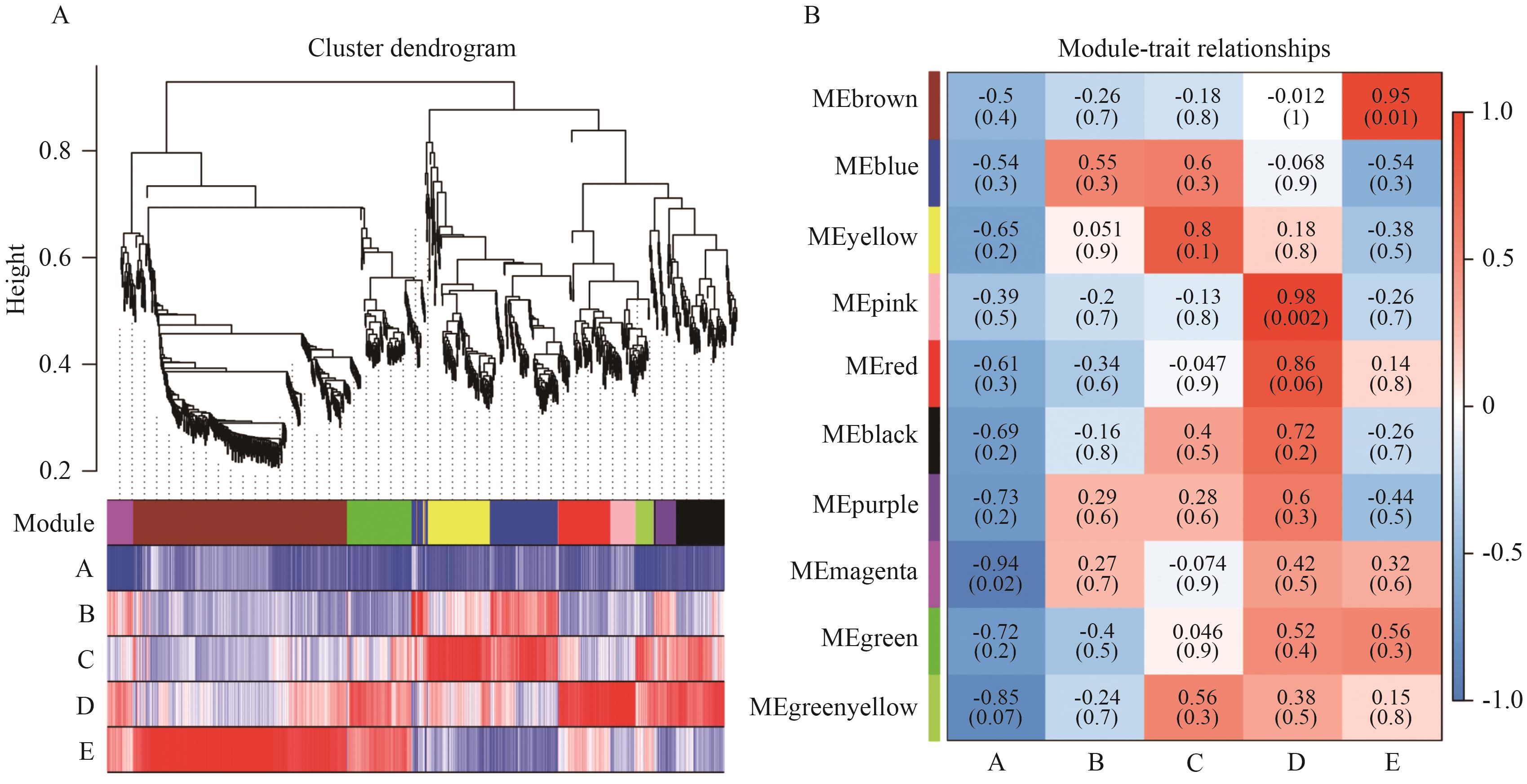
图3 加权基因共表达网络分析A:系统聚类树和模块划分图;B:共表达基因模块与性状的相关性热图
Fig. 3 Analysis of WGCNAA: Hierarchical clustering tree and module division diagram. B: Correlation heatmap between co-expressed gene modules and traits
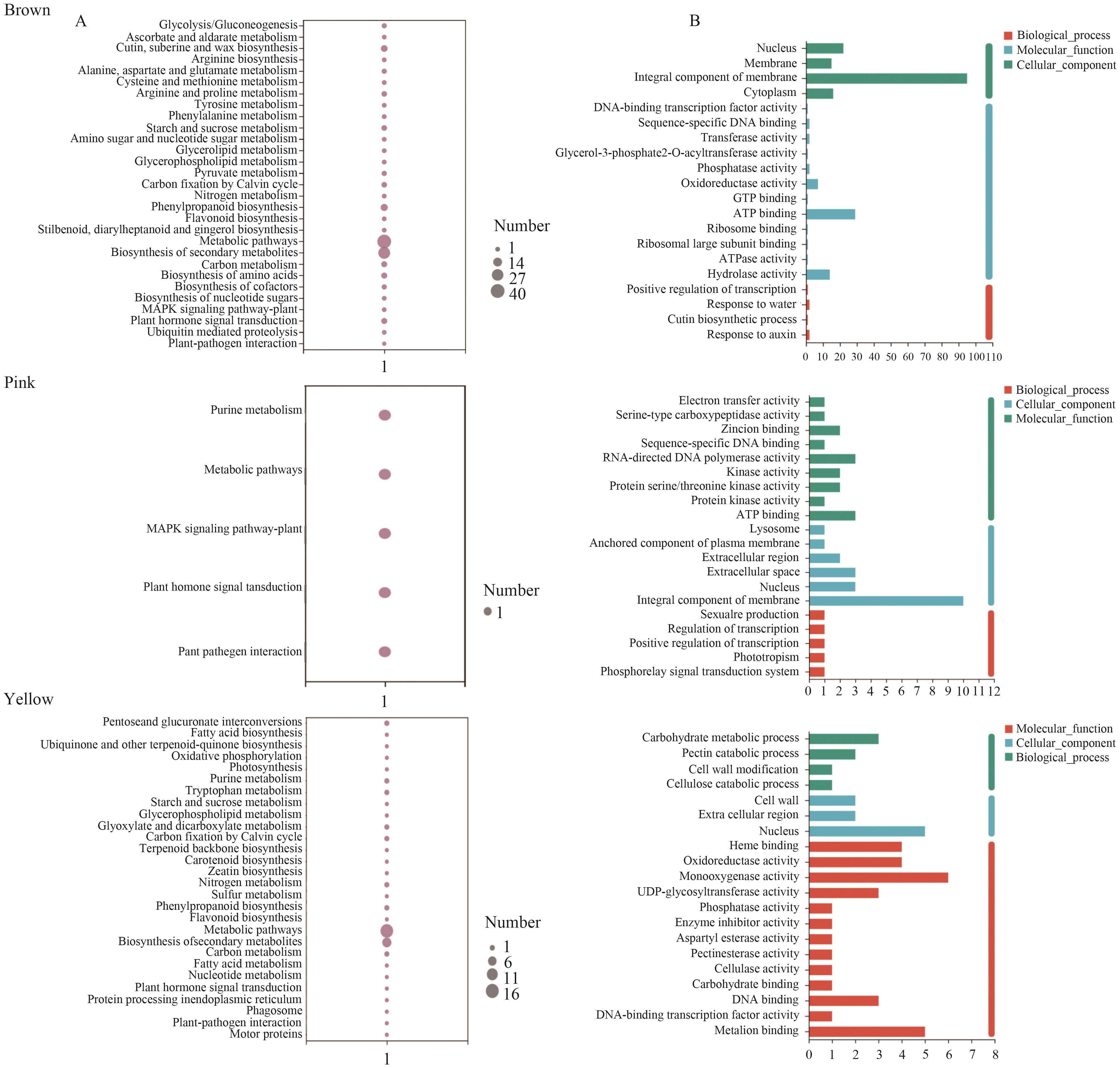
图4 花花柴耐盐特异性模块富集分析A:花花柴耐盐特异性模块KEGG富集分析;B:花花柴耐盐特异性模块GO富集分析
Fig. 4 Enrichment analysis of specific modules in the tolerance of K. caspia to saltA: KEGG enrichment analysis of specific modules for salt-tolerance in Karelinia caspia. B: GO enrichment analysis of specific modules to salt-tolerance in K. caspia

图5 模块核心基因互作网络图及各时期表达热图
Fig. 5 Interaction network of hub genes in modules and heatmaps of their expressions during different stages
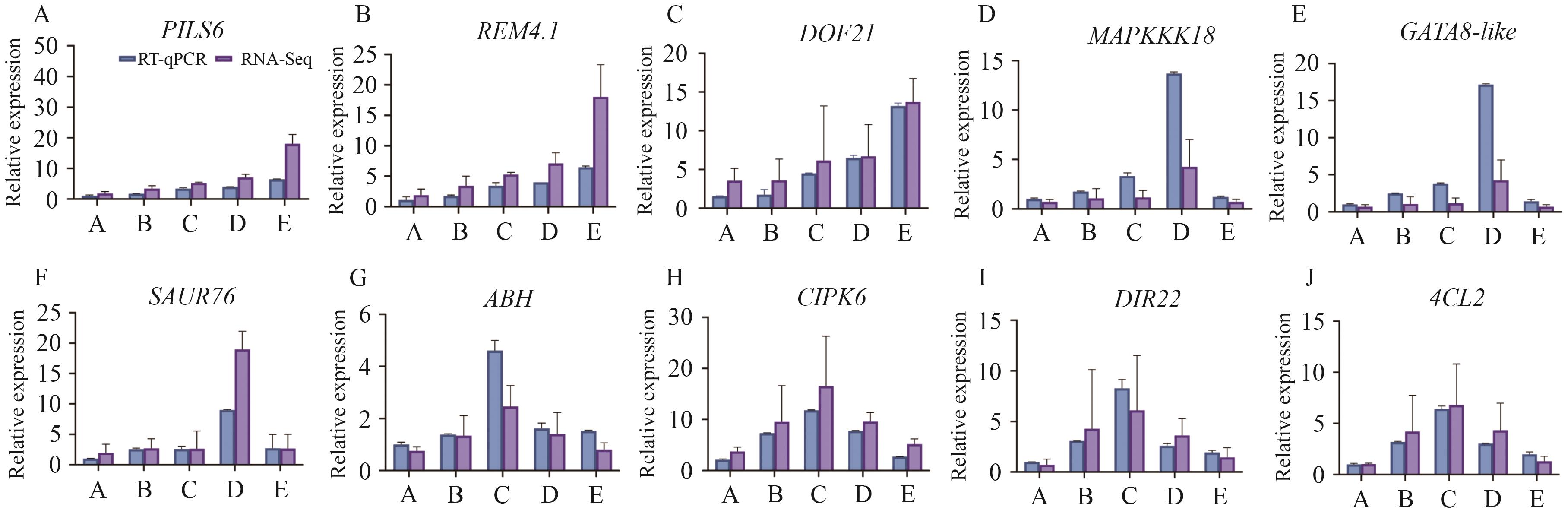
图6 核心基因的 RT-qPCR 验证
Fig. 6 RT-qPCR validation of hub genes
| [1] | van Zelm E, Zhang YX, Testerink C. Salt tolerance mechanisms of plants [J]. Annu Rev Plant Biol, 2020, 71: 403-433. |
| [2] | Guo Q, Han JW, Li C, et al. Defining key metabolic roles in osmotic adjustment and ROS homeostasis in the recretohalophyte Karelinia caspia under salt stress [J]. Physiol Plant, 2022, 174(2): e13663. |
| [3] | 刘晨, 徐浩博, 斯钰阳, 等. 基于转录组学的植物响应盐胁迫调控机制研究进展 [J]. 浙江农业学报, 2022, 34(4): 870-878. |
| Liu C, Xu HB, Si YY, et al. Research progress on regulation mechanism of plant response to salt stress based on transcriptomics [J]. Acta Agric Zhejiangensis, 2022, 34(4): 870-878. | |
| [4] | Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis [J]. BMC Bioinformatics, 2008, 9: 559. |
| [5] | Zhen X, Liu XY, Zhang XM, et al. Identification of core genes involved in the response of Apocynum venetum to salt stress based on transcriptome sequencing and WGCNA [J]. PLoS One, 2024, 19(4): e0300277. |
| [6] | 肖胜华, 陆妍, 李安子, 等.棉花AP2/ERF转录因子GhTINY2负调控植株抗盐性的功能分析[J].作物学报, 2024, 50(1):126-137. |
| Xiao SG, Lu Y, Li AZ, et al. Functional analysis of the negative regulation of the AP2/ERF transcription factor GhTINY2 on salt stress tolerance in cotton [J]. Acta Agron Sin, 2024, 50(1):126-137. | |
| [7] | 李旭凯, 李任建, 张宝俊. 利用WGCNA鉴定非生物胁迫相关基因共表达网络 [J]. 作物学报, 2019, 45(9): 1349-1364. |
| Li XK, Li RJ, Zhang BJ. Identification of rice stress-related gene co-expression modules by WGCNA [J]. Acta Agron Sin, 2019, 45(9): 1349-1364. | |
| [8] | 张毅, 吴万亿, 刘霞宇, 等. 利用WGCNA鉴定甘薯耐盐相关共表达网络及核心基因 [J]. 河南农业科学, 2021, 50(6): 16-27. |
| Zhang Y, Wu WY, Liu XY, et al. Identification of salt tolerance co-expression modules and hub genes in Ipomoea batatas by WGCNA [J]. J Henan Agric Sci, 2021, 50(6): 16-27. | |
| [9] | 张晨, 孟林, 毛培春, 等. 盐生植物花花柴KcSOS1基因RNA干扰遗传转化体系构建及其功能初步鉴定 [J]. 植物生理学报, 2020, 56(3): 529-539. |
| Zhang C, Meng L, Mao PC, et al. Genetic transformation system construction of KcSOS1-RNAi and primary functional identification in halophyte Karelinia caspia [J]. Plant Physiol J, 2020, 56(3): 529-539. | |
| [10] | Hu TY, Chen JW, Lin XQ, et al. Comparison of the DNBSEQ platform and Illumina HiSeq 2000 for bacterial genome assembly [J]. Sci Rep, 2024, 14(1): 1292. |
| [11] | 田润萌. 利用RNA-seq和WGCNA挖掘黄秋葵品质性状相关基因 [D]. 福州: 福建农林大学, 2023. |
| Tian RM. Mining genes related to quality traits of okra by RNA-seq and WGCNA [D]. Fuzhou: Fujian Agriculture and Forestry University, 2023. | |
| [12] | Haas BJ, Papanicolaou A, Yassour M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis [J]. Nat Protoc, 2013, 8(8): 1494-1512. |
| [13] | Seppey M, Manni M, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness [J]. Methods Mol Biol, 2019, 1962: 227-245. |
| [14] | Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome [J]. BMC Bioinformatics, 2011, 12: 323. |
| [15] | Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2 [J]. Genome Biol, 2014, 15(12): 550. |
| [16] | Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks [J]. Nat Protoc, 2012, 7(3): 562-578. |
| [17] | Roumeliotis E, Kloosterman B, Oortwijn M, et al. The effects of auxin and strigolactones on Tuber initiation and stolon architecture in potato [J]. J Exp Bot, 2012, 63(12): 4539-4547. |
| [18] | Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks [J]. Genome Res, 2003, 13(11): 2498-2504. |
| [19] | Chen CJ, Chen H, Zhang Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data [J]. Mol Plant, 2020, 13(8): 1194-1202. |
| [20] | Guo Q, Meng L, Han JW, et al. SOS1 is a key systemic regulator of salt secretion and K+/Na+ homeostasis in the recretohalophyte Karelinia caspia [J]. Environ Exp Bot, 2020, 177: 104098. |
| [21] | Xie GW, Zou XZ, Liang ZS, et al. GBF family member PfGBF3 and NAC family member PfNAC2 regulate rosmarinic acid biosynthesis under high light [J]. Plant Physiol, 2024, 195(2): 1728-1744. |
| [22] | Le L, Guo WJ, Du DY, et al. A spatiotemporal transcriptomic network dynamically modulates stalk development in maize [J]. Plant Biotechnol J, 2022, 20(12): 2313-2331. |
| [23] | 王瀚祥, 李广存, 徐建飞, 等. 植物耐盐机理研究进展 [J]. 作物杂志, 2022, (5):1-12. |
| Wang HX, Li GC, Xu JF, et al. Research progress on plant salt tolerance mechanisms [J]. Crops, 2022, (5):1-12. | |
| [24] | Wang D, Yang N, Zhang CY, et al. Transcriptome analysis reveals molecular mechanisms underlying salt tolerance in halophyte Sesuvium portulacastrum [J]. Front Plant Sci, 2022, 13: 973419. |
| [25] | Zhu ZH, Zhou YQ, Liu XY, et al. Integrated transcriptomic and metabolomic analyses uncover the key pathways of Limonium bicolor in response to salt stress [J]. Plant Biotechnol J, 2025, 23(3): 715-730. |
| [26] | Li C, Mur LAJ, Wang QH, et al. ROS scavenging and ion homeostasis is required for the adaptation of halophyte Karelinia caspia to high salinity [J]. Front Plant Sci, 2022, 13: 979956. |
| [27] | 杨金凤. 柽柳耐盐特性的转录组和代谢组分析 [D]. 泰安: 山东农业大学, 2024. |
| Yang JF. Transcriptome and metabonomic analysis of salt tolerance of Tamarix chinensis [D]. Tai’an: Shandong Agricultural University, 2024. | |
| [28] | Liu JN, Fang HC, Liang Q, et al. Genomic analyses provide insights into the evolution and salinity adaptation of halophyte Tamarix chinensis [J]. GigaScience, 2022, 12: giad053. |
| [29] | Kumar K, Raina SK, Sultan SM. Arabidopsis MAPK signaling pathways and their cross talks in abiotic stress response [J]. J Plant Biochem Biotechnol, 2020, 29(4): 700-714. |
| [30] | Tripathi V, Parasuraman B, Laxmi A, et al. CIPK6, a CBL-interacting protein kinase is required for development and salt tolerance in plants [J]. Plant J, 2009, 58(5): 778-790. |
| [31] | Zou XM, Sun HM. DOF transcription factors: Specific regulators of plant biological processes [J]. Front Plant Sci, 2023, 14: 1044918. |
| [32] | Nutan KK, Singla-Pareek SL, Pareek A. The Saltol QTL-localized transcription factor OsGATA8 plays an important role in stress tolerance and seed development in Arabidopsis and rice [J]. J Exp Bot, 2020, 71(2): 684-698. |
| [33] | 殷爱萍, 李振, 刘翀, 等.烟草PILS6基因的生物信息学与表达模式分析[J].生物过程, 2024, 14(4): 202-209. |
| Yin AP, Li Z, Liu C, et al. Bioinformatics and expression pattern analysis of the PILS6 gene in tobacco (Nicotiana tabacum) [J]. Bioprocess, 2024, 14(4):202-209. | |
| [34] | 刘超逸, 王宇航. 植物生长素响应基因SAUR研究进展 [J]. 中国农学通报, 2024, 40(18): 83-89. |
| Liu CY, Wang YH. Plant auxin responsive gene SAUR a review [J]. Chin Agric Sci Bull, 2024, 40(18): 83-89. | |
| [35] | Gui JS, Zheng S, Liu C, et al. OsREM4.1 interacts with OsSERK1 to coordinate the interlinking between abscisic acid and brassinosteroid signaling in rice [J]. Dev Cell, 2016, 38(2): 201-213. |
| [36] | 张会龙, 武霞, 尧俊, 等. 胡杨PeREM1.3过表达提高烟草耐盐性的机制 [J]. 北京林业大学学报, 2019, 41(1): 1-9. |
| Zhang HL, Wu X, Yao J, et al. Overexpression mechanism of PeREM1.3 from Populus euphratica enhancing salt tolerance in transgenic tobacco [J]. J Beijing For Univ, 2019, 41(1): 1-9. | |
| [37] | Mindrebo JT, Nartey CM, Seto Y, et al. Unveiling the functional diversity of the alpha/beta hydrolase superfamily in the plant Kingdom [J]. Curr Opin Struct Biol, 2016, 41: 233-246. |
| [38] | Davin LB, Lewis NG. Dirigent proteins and dirigent sites explain the mystery of specificity of radical precursor coupling in lignan and lignin biosynthesis [J]. Plant Physiol, 2000, 123(2): 453-462. |
| [39] | Li NH, Zhao M, Liu TF, et al. A novel soybean dirigent gene GmDIR22 contributes to promotion of lignan biosynthesis and enhances resistance to Phytophthora sojae [J]. Front Plant Sci, 2017, 8: 1185. |
| [40] | 田晓明, 颜立红, 向光锋, 等. 植物4香豆酸: 辅酶A连接酶研究进展 [J]. 生物技术通报, 2017, 33(4): 19-26. |
| Tian XM, Yan LH, Xiang GF, et al. Research progress on 4-coumarate: coenzyme a ligase(4CL) in plants [J]. Biotechnol Bull, 2017, 33(4): 19-26. | |
| [41] | Ma JY, Zuo DJ, Zhang XD, et al. Genome-wide identification analysis of the 4-coumarate: CoA ligase (4CL) gene family expression profiles in Juglans regia and its wild relatives J. mandshurica resistance and salt stress [J]. BMC Plant Biol, 2024, 24(1): 211. |
| [1] | 刘泽洲, 段乃彬, 岳丽昕, 王清华, 姚行浩, 高莉敏, 孔素萍. 大蒜叶片蜡质成分分析及蜡质缺失基因Ggl-1筛选[J]. 生物技术通报, 2025, 41(9): 219-231. |
| [2] | 闫梦阳, 梁晓阳, 戴君昂, 张妍, 关团, 张辉, 刘良波, 孙志华. 阿莫西林降解菌的筛选及降解机制研究[J]. 生物技术通报, 2025, 41(9): 314-325. |
| [3] | 朱丽娟, 张锴, 温晓蕾, 褚佳豪, 史凤玉, 王艳丽. 基于WGCNA挖掘野生大豆耐镉关键基因[J]. 生物技术通报, 2025, 41(8): 124-136. |
| [4] | 王月琛, 韩鑫骐, 魏文敏, 崔兆兰, 罗阳美, 陈鹏如, 王海岗, 刘龙龙, 张莉, 王纶. 黍稷落粒的生物学基础研究及落粒调控基因的鉴定[J]. 生物技术通报, 2025, 41(7): 164-171. |
| [5] | 郭秀娟, 冯宇, 吴瑞香, 王利琴, 杨建春. Ca2+处理对胡麻种子萌发影响的转录组分析[J]. 生物技术通报, 2025, 41(7): 139-149. |
| [6] | 李小欢, 陈相宇, 陶麒宇, 朱玲, 唐铭, 姚银安, 汪丽君. PtoMYB61对毛白杨木质素合成及耐盐性的影响[J]. 生物技术通报, 2025, 41(6): 284-296. |
| [7] | 李旭娟, 李纯佳, 刘洪博, 徐超华, 林秀琴, 陆鑫, 刘新龙. 甘蔗腋芽形成发育过程的转录组分析[J]. 生物技术通报, 2025, 41(3): 202-218. |
| [8] | 岳丽昕, 王清华, 刘泽洲, 孔素萍, 高莉敏. 基于转录组和WGCNA筛选大葱雄性不育相关基因[J]. 生物技术通报, 2024, 40(9): 212-224. |
| [9] | 赵海平, 刘林, 王昕璐, 岳鹏飞, 孔维军, 王蒙. 基于WGCNA探讨葛根素对呕吐毒素诱导C6细胞损伤的保护作用[J]. 生物技术通报, 2024, 40(9): 301-310. |
| [10] | 高萌萌, 赵天宇, 焦馨悦, 林春晶, 关哲允, 丁孝羊, 孙妍妍, 张春宝. 大豆细胞质雄性不育系及其恢复系的比较转录组分析[J]. 生物技术通报, 2024, 40(7): 137-149. |
| [11] | 白志元, 徐菲, 杨午, 王明贵, 杨玉花, 张海平, 张瑞军. 大豆细胞质雄性不育弱恢复型杂种F1育性转变的转录组分析[J]. 生物技术通报, 2024, 40(6): 134-142. |
| [12] | 秦健, 李振月, 何浪, 李俊玲, 张昊, 杜荣. 肌源性细胞分化的单细胞转录谱变化及细胞间通讯分析[J]. 生物技术通报, 2024, 40(6): 330-342. |
| [13] | 杨淇, 魏子迪, 宋娟, 童堃, 杨柳, 王佳涵, 刘海燕, 栾维江, 马轩. 水稻组蛋白H1三突变体的创建和转录组学分析[J]. 生物技术通报, 2024, 40(4): 85-96. |
| [14] | 赵锐, 狄靖宜, 张广通, 刘浩, 高伟霞. 基于转录组学挖掘兽疫链球菌内源表达元件及高产透明质酸应用[J]. 生物技术通报, 2024, 40(10): 296-304. |
| [15] | 韩志阳, 贾子苗, 梁秋菊, 王轲, 唐华丽, 叶兴国, 张双喜. 二套小麦-簇毛麦染色体附加系苗期耐盐性及籽粒硒和叶酸的含量[J]. 生物技术通报, 2023, 39(8): 185-193. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||